
Abstract
Soft tissue type perineuriomas (STP), or Extraneural perineuriomas, are typically found in the superficial extremities or trunk of adult patients. Their incidence in the head and neck is exceptionally rare, particularly amongst the pediatric population. Since 1978, only 19 cases of pediatric STP have been reported, with only one in the neck. This case report describes the second case of STP in the neck of a child as well as reviews the current literature on pediatric STP. The pattern of patient genetic anomalies associated with the few pediatric STP cases encountered suggests an association between genetic aberrations and STP. Clinicians should be aware of STP when formulating a differential diagnosis of pediatric soft tissue masses in the head and neck despite the rarity of this tumor.
Introduction
First described in 1978 by Lazarus and Trombetta, perineuriomas are rare, typically benign, peripheral nerve sheath tumors. 1 Historically, these neoplasms represented a challenging diagnostic entity until the advent of immunohistochemical (IHC) techniques which allowed for better characterization of their pathologic features and hence facilitated diagnosis. Composed of well-differentiated perineural cells, four variants of perineurioma are recognized—Intraneural, Reticular, Sclerosing, and Soft tissue. 2 Soft tissue type perineuriomas (STP), or Extraneural perineuriomas, are typically found in the superficial extremities or trunk of adult patients. 3 Soft tissue type perineuriomas in the head and neck are exceptionally rare, particularly amongst the pediatric population, and as such the natural history of STP in the pediatric population is assumed based on data from case reports of adults with similar neoplasms.
Herein, we illustrate a new case of a pediatric STP in the cervical soft tissues, only the second such case described in the literature, as well as present the clinical, histologic, and IHC characteristics of these lesions. Concurrently, we contrast the IHC characteristics of STP with other rare pediatric nerve tumors, including other variants of perineurioma. We also describe the genetic aberrations potentially associated with pediatric STPs. This report serves to better clarify the current understanding of these uncommon lesions to assist providers when faced with diagnosis and treatment of STP.
Case Report
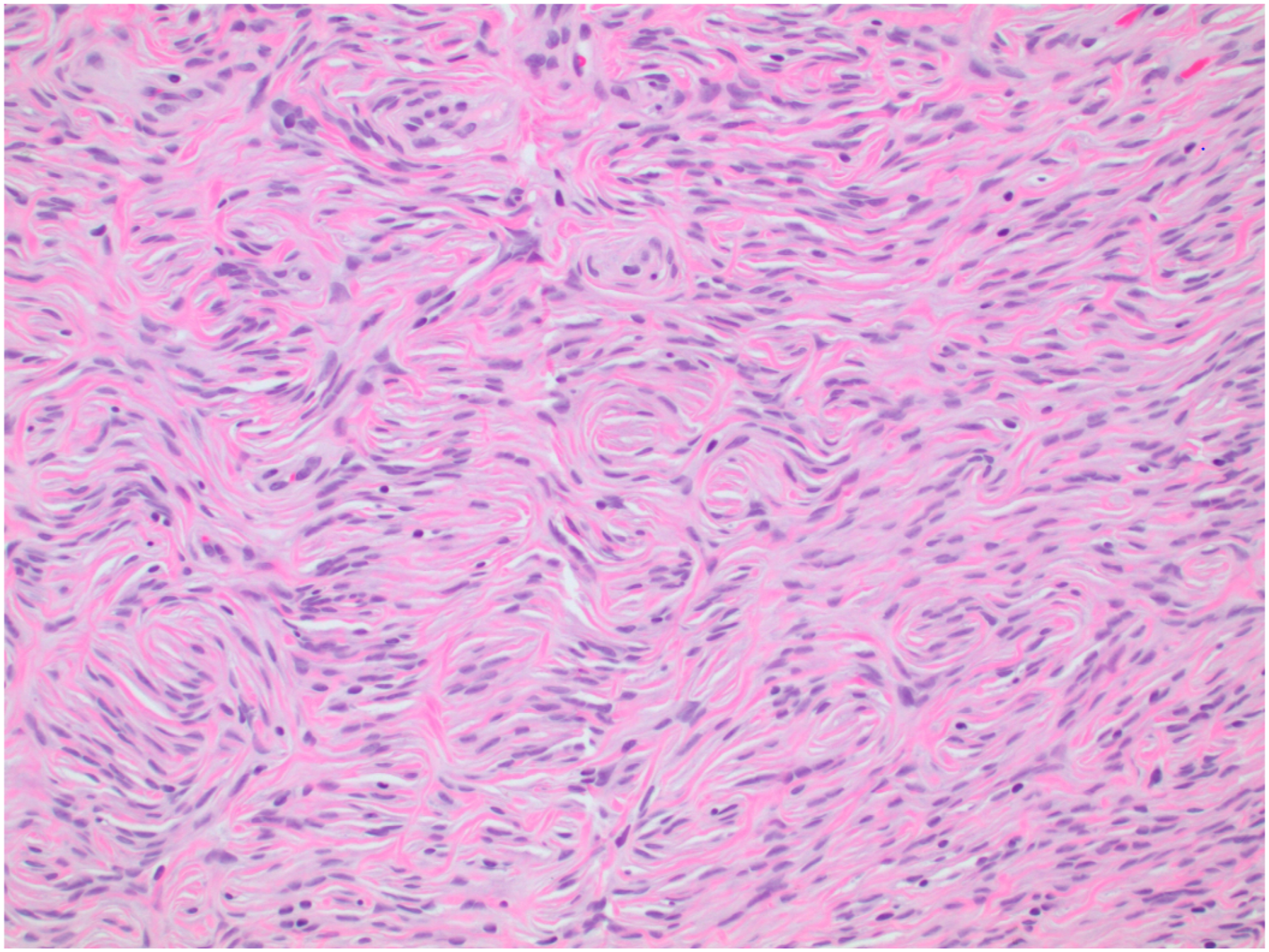
A 15-year-old male with no previous medical history presented to our Otolaryngology—Head & Neck Surgery practice for evaluation of a gradually enlarging, right-sided neck mass. The patient first noticed the mass 10 months prior to his office visit and reported non-fluctuating progressive growth over time. He denied any local pain, sensory changes, or systemic symptoms including absence of fevers, night sweats, or weight loss. Physical examination demonstrated a mobile, non-tender 2.0 cm mass at level IB in the right neck adjacent to the inferior border of the mandible. Superficial ultrasound demonstrated a well-circumscribed, hypoechoic, homogenous mass in the subcutaneous plane. Magnetic resonance imaging demonstrated a well-defined ovoid mass superficial to the platysma and the superficial muscular aponeurotic system distinct from the submandibular gland (Figure 1). The lesion was isointense to muscle on T1-weighted sequences, mildly hyperintense on T2-weighted fat-suppressed sequences, and enhanced following gadolinium contrast administration. Subsequent ultrasound-guided fine-needle aspiration yielded equivocal findings demonstrating a variety of spindle cells and rare lymphocytes admixed in a matrix of mucoid stroma. The decision was made to proceed with excision of the mass for diagnostic and potentially curative purposes. The patient underwent successful and uncomplicated surgical extirpation of this tumor. Pathologic evaluation revealed a well-circumscribed neoplasm composed of bland spindle cells arranged in a storiform pattern (Figures 2). Fluorescence in-situ hybridization (FISH) for 22q13 rearrangement of the PDGFB gene was performed to rule out dermatofibrosarcoma protuberans, another rare soft tissue tumor with unpredictable subclinical extension. The results of this analysis were negative. Based on these findings, in conjunction with the clinical and radiographic features of this lesion, the diagnosis of STP was made. Axial plane Magnetic resonance imaging of the neck without contrast demonstrating a well-defined, ovoid heterogeneously mildly T2 hyperintense mass. Storiform pattern of pathologic specimen, H&E stain, 20x.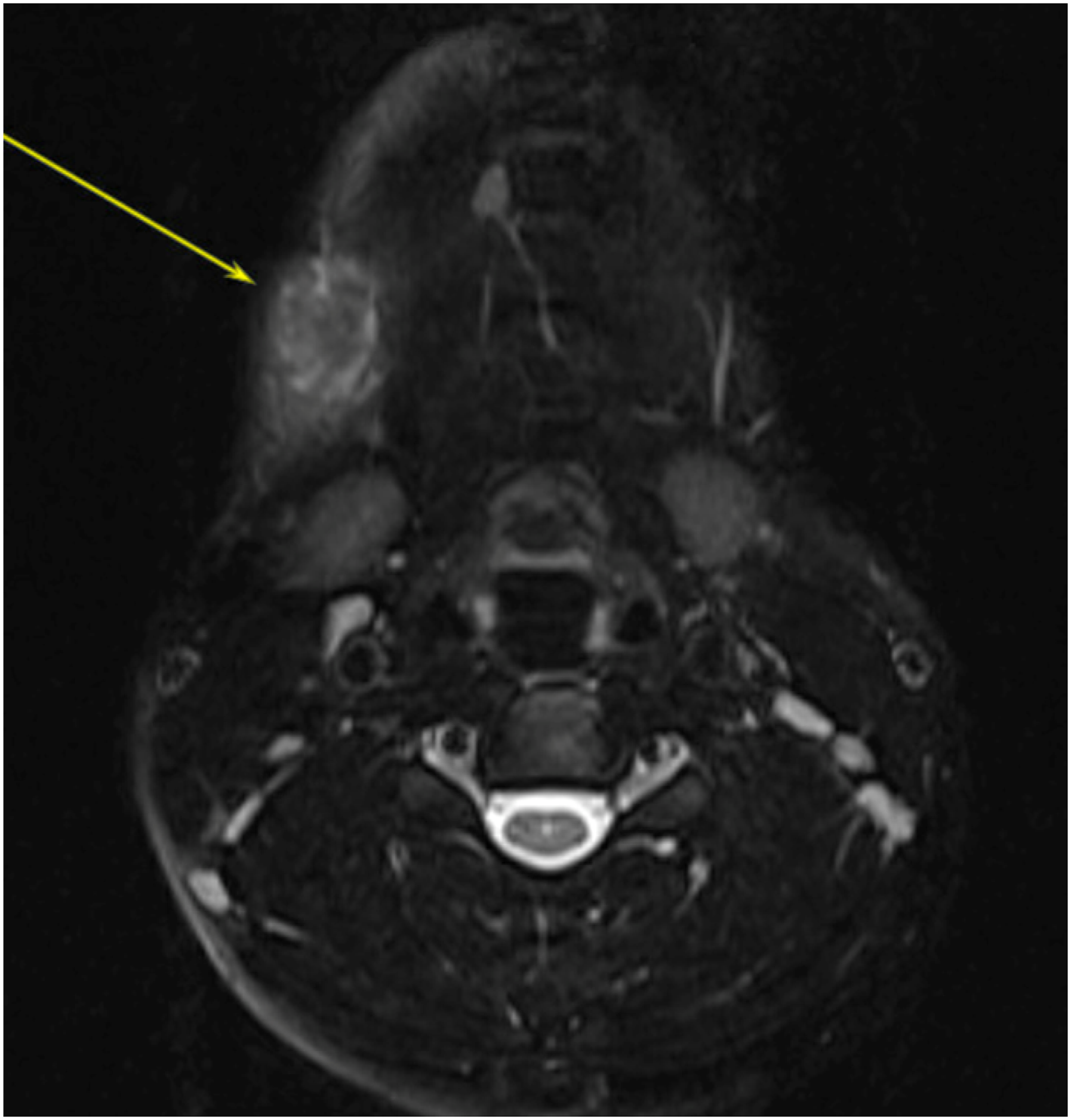
Discussion
Perineuriomas commonly have a benign course and rarely recur after surgical resection. Despite the excellent prognosis associated with these lesions, presence of these tumors may indicate an underlying genetic abnormality given that perineuromas are frequently associated with a chromosomal anomaly in the affected patient. Such an association has been discussed in a wide range of reports; however, due to the low incidence of these lesions in the general population, the correlation between genetic anomalies and soft tissue perineuriomas has been difficult to qualify. Many intra- and extra-neural perineuriomas are associated with mutation in chromosome 22. 4 In a case series by Carter et al, 13 out of 14 STPs were associated with at least two chromosomal abnormalities, most commonly chr22q deletions of the NF2 locus (n = 6) and chr17q deletions of the NF1 locus (n = 4). 5 There have also been reports of NF2 point mutations in perineuriomas at the 5’-untranslated region and within exons of the locus. 6 Association with neurofibromatosis type 1 (NF1) has been documented in one case of an adult male with a soft tissue perineurioma in the leg. 7 Additionally, a soft tissue perineurioma is described in Al-Adnani et al, in a 10-year-old male with NF1. 8 The link between chr22, implicated in NF2 (neurofibromatosis type 2), and chr17, implicated in NF1, and the subsequent development of soft tissue perineuriomas is as yet not fully clear. Al-Adnani et al, remarks that 50% of malignant peripheral nerve sheath tumors (MPNST) are associated with NF1. Furthermore, patients with NF1 and NF2 are more likely to develop other rare tumors of neural origin such as hybrid peripheral nerve sheath tumors or Schwannomas.
We could not rule out the presence of additional chromosomal abnormalities in our patient without further testing; however, his family opted out of additional genetic analysis due to financial barriers in confluence with limited evidence supporting significant prognostic improvement in early detection and treatment of chromosomal abnormality comorbidities. It should be noted that isolated reports of cytogenetic anomalies have indeed been discovered in cases of STP such as a 10q24 rearrangement in a case of STP in the foot 9 and a chr13 deletion in an STP of the thigh. 10 Balarezo et al report a complex translocation between chromosomes 2, 4, and 9 that involves the ABL1 proto-oncogene in association with STP. 11 Brock et al. reports a translocation between chromosomes 8 and 9 in a patient with an intra-abdominal STP. 12 Koutlas et al report the only pediatric STP within the head and neck that is associated with a genetic syndrome—Gorlin syndrome (a.k.a nevoid basal cell carcinoma syndrome). 13
Conclusion
Pediatric STPs in the head and neck are rare lesions and their association with concomitant genetic anomalies is poorly understood. Our report demonstrates only the second documented case of a soft tissue perineurioma of the neck in a pediatric patient. The pattern of genetic anomalies amongst the few pediatric STP cases encountered suggests an association between genetic aberrations and STP in some cases; however the cost of genotyping studies, coupled with a lack of sample size due to infrequency of these lesions, has led to limited knowledge of concomitant genetic disease development. The opportunity exists for further study of chromosomal anomalies associated with STP as new cases of STP are diagnosed.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Written consent was obtained by the patients’ parents prior to the start of all research activity.