
Abstract
Glial choristomas are isolated masses of mature brain tissue that are found outside the spinal cord or cranial cavity. These masses are rare, especially in the middle ear. We describe the case of an 81-year-old man who presented with left-sided chronic otitis media, mastoiditis, hearingloss, tinnitus, and aural fullness. He was found to have a glial choristoma of the middle ear on the left. Otologic surgeons should be aware of the possibility of finding such a mass in the middle ear and be familiar with the differences in treatment between glial choristomas and the more common encephaloceles.
Introduction
Opacification within the middle ear is a common finding on radiologic imaging. It can be caused by inflammatory changes, cholesteatomas, carcinomas, and other common pathologies. Middle ear opacification can also be caused by more unusual entities, such as teratomas, central nervous system (CNS) neoplasms, vascular tumors, and other lesions.
Middle ear lesions are associated with increased risks of cerebrospinal fluid leak and meningitis. In such cases, otologic surgeons should be prepared to perform a combined otologic and neurotologic (intracranial) procedure to repair dural and brain floor defects when necessary.
We report a case of glial choristoma of the middle ear, and we review the literature on the nature and management of this lesion in this location.
Case Report
An 81 -year-old man presented for evaluation of chronic otitis media, mastoiditis, diminished hearing, tinnitus, and left aural fullness of several months’ duration. The appearance of both tympanic membranes was normal, and no evidence of effusion was noted. Tympanometry revealed decreased compliance in the left middle ear, and audiometry demonstrated a mixed hearing loss bilaterally. A myringotomy tube was placed in the left ear, but the patient continued to experience persistent purulent otorrhea despite medical therapy.
Computed tomography (CT) of the temporal bones revealed opacification of the left mastoid process that appeared to represent either middle ear fluid or soft tissue (figure 1). CT also showed intact ossicles, minimal erosion of the scutum, an absence of air-cell coalescence, and possible erosion of the attic with left middle fossa communication. Magnetic resonance imaging (MRI) of the brainstem and internal auditory canals demonstrated a large amount of left middle ear and mastoid opacification consistent with inflammatory tissue.
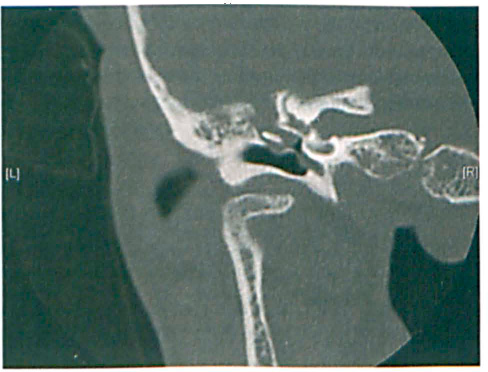
CT of the left temporal bone shows the opacity in the left middle ear and the possible erosion of the tegmen tympani above the attic. The ossicles are intact.
The patient was taken to the operating room for a left tympanomastoidectomy. During the procedure, the middle ear was found to be filled with fluid. When the fluid was suctioned, a smooth mass was found to be filling the upper portion of the middle ear medial to the incus and in continuity with the horizontal portion of the facial nerve. The stapes had been partially displaced inferiorly. Palpation of the ossicles revealed that the malleus and incus were mobile, the attachment between the stapes and incus was somewhat loose, and the stapes footplate was fixed.
After a complete intact-canal-wall tympanomastoidectomy, the mass was seen to extend inferiorly; it involved the chorda tympani, but it did not extend any farther into the mastoid than the antrum. The mass was separated from the facial nerve and the undersurface of the ossicles with the use of a facial nerve stimulator and angled instruments, and it was resected in its entirety. The lesion had extended from slightly below the level of the pyramidal process to just anterior to the cochleariform process; it did not extend below the level of the umbo.
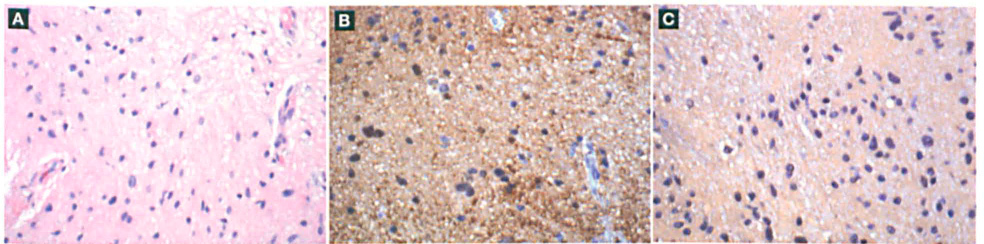
Hematoxylin and eosin staining revealed disorganized glia-like tissue on a loosely fibrillary background (figure 2, A). The sections were stained for S-100 protein, glial fibrillary acidic protein (GFAP), cytokeratin, and microtubule-associated protein. Stains were positive for S-100 protein (figure 2, B) and GFAP (figure 2, C). After correlating the radiologic and operative findings and obtaining surgical confirmation of the absence of a cranial connection, we diagnosed the lesion as a middle ear glial choristoma.
Discussion
A glial choristoma is a mass-like lesion of mature brain tissue that is found outside the spinal cord and cranial cavity. 1 Approximately 40 cases have been reported in the middle ear.1–19 These collections of heterotopic brain tissue are usually found in extracranial midline structures—typically the nose, nasopharynx (nasal glioma), oropharynx, orbits, skin, soft palate, tonsils, tongue, lip, and submandibular area. They have been found as far caudally as the lungs and peritoneum.2,3
Glial choristomas are classified into six types according to their location and possible pathologic mechanisms:
intraparenchymal lesions;
dural and leptomeningeal lesions;
intracranial extracerebral lesions;
midline lesions;
distal lesions of the lung and uterus; and
The salivary glands are the most common source of middle ear choristomas. 6 The presence of heterotopic brain tissue at sites away from the midline is uncommon, and therefore glial choristomas of the middle ear are rare.
The pathology of glial choristoma is not completely clear. A leading hypothesis suggests that glial choristomas are variants of an encephalocele with a detached or absorbed pedicle that had once connected the glial tissue directly to a ventricular or subarachnoid area. 1 Heterotopic brain tissue may become trapped in an unusual location if an imperfection or delay in a fetus's skull development allows for tissue projection.1,7,8
Other cases have no connection to the CNS; instead, the connection may be provided by a small congenital defect in the tegmen tympani or overlying temporal bone. For example, Uğruz et al described a case in which a bony defect was present at the tegmen tympani, but the dura remained intact, so there was no connection to the CNS. 9
Small defects in the tegmen tympani have been seen in as many as 20% of autopsy cases. 10 These lesions had no true connection to the CNS; instead, it might have been a congenital defect that allowed the brain tissue to enter the middle ear. Additionally, the closure of cranial coverings may be inhibited by an overgrowth of the neural tube, which results in the creation of pockets of tissue. 1
Salivary gland choristomas of the middle ear are often associated with abnormalities in the facial nerve and branchial clefts, which suggests the role of developmental errors. 6 Other theories suggest (1) the differentiation of displaced pluripotential neuroectodermal cells into mature brain tissue and (2) detachment of extracranial embryonic tissue independent of cranial closure. 7
In support of the theory of an encephalocele variant, some reports have suggested an association between a middle ear glial choristoma and a previous infection, inflammatory process, trauma, or surgery that exposed the CNS tissue.3,10 Cholesteatomas were described in 3 cases with heterotopic brain tissue, which suggests that glial choristomas can be caused by local destruction of bony segments subsequent to cholesteatoma, meningitis, or otitis media and that the cholesteatomas can occasionally expand into the intracranial cavity. 3 However, other authors have reported none of these predisposing factors, which perhaps separates these true lesions from the glial variants of acquired encephaloceles.9,11 Nevertheless, one cannot rule out an unrecognized trauma, an unidentified inflammatory process, or another unforeseen factor. Also, as mentioned previously, small congenital defects may not be recognized.
Histologically, glial choristomas are composed of mature CNS components, including glial cells and neural cells. Other histologic findings can include ependymal cells, choroid plexus papillary projections, ganglion cells, oligodendrocytes, spongioblasts, and pigment epithelium. 12 The presence or absence of neural cells may depend on the stage when the glial tissue separated from its developmental embryonal tissue. 13 In brain development, neuronal precursors emerge during the 10th week of intrauterine life. Therefore, no neuronal elements would be seen in the heterotopic tissue if it separated before this crucial time. 13 Examination of the pedicle of the mass may also help determine its pathology.
CT is valuable for assessing possible bony involvement, although high-resolution CT can miss bone abnormalities, as was seen in 8 cases of spontaneous encephalocele that were later found to involve bone and dural dehiscence during surgical exploration. 1 CT might also fail to define abnormal tissue densities, but it can intermittently reveal a translucency or a mass in the middle ear. 1 MRI can show a connection between the tissue and the adjoining brain; on T1 - and T2-weighted imaging, the appearance of a glial choristoma is similar to that of brain tissue. Of note, dys-plastic neural tissue may cause a glial choristoma to appear hyperintense on T2-weighted MRI. 7
Correlation with radiologic and surgical findings is important in differentiating heterotopic brain tissue from an encephalocele, as there is no difference between the two on histology. It is also important to differentiate the two in order to avoid postoperative recurrent infections in patients with middle ear encephalocele. 6 For glial choristomas, complete excision provides definitive treatment, while encephaloceles might require additional surgical measures in order to repair dural defects.
Although they are rare, glial choristomas of the middle ear should be considered in the differential diagnosis of a patient with a middle ear mass. Because no significant histologic differences exist between heterotopic brain tissue and encephalocele, it is important to distinguish between the two clinically in order to plan appropriate surgery. A combination of clinical suspicion, imaging, and intraop erative attentiveness is needed to optimize treatment and prevent serious complications.