
Abstract
Juvenile xanthogranuloma (JXG) is a benign, non-Langerhans cell histiocytic lesion that generally affects infants and children. These lesions characteristically appear as a solitary, yellow, cutaneous nodule of the head, neck, or trunk. Subcutaneous and extracutaneous forms can involve the gastrointestinal tract, kidney, lung, gonads, pericardium, central nervous system, temporal bone, larynx, and eye. We describe the clinical presentation, imaging, histochemical findings, and management of a solitary JXG of the tympanic membrane in a 17-month-old girl. The patient underwent surgical resection and was without disease several months following surgery and reconstruction of the defect. To the best of our knowledge, this is the first reported case of a JXG of the tympanic membrane.
Introduction
Juvenile xanthogranuloma (JXG) is a benign, non-Langerhans cell histiocytic lesion that is most commonly found in or on the skin of the head, neck, or trunk. It was first described by Adamson in 1905 as congenital xanthoma multiplex. 1 In 1912, McDonagh renamed these lesions naevo-xantho-endotheliomata. 2 In 1954, Helwig and Hackney called the condition xanthogranuloma based on its histologic appearance. 3
To the best of our knowledge, no case of JXG of the tympanic membrane has been previously reported in the literature. We describe the clinical, imaging, surgical, and histochemical features of such a case, as well as our management of it.
Case report
A 17-month-old girl was referred to us for evaluation of recurrent left-sided otitis media. Physical examination revealed that she had a bulging lesion in the posterosuperior quadrant of the left tympanic membrane. After further examination under local anesthesia, tympanostomy tube placement was carried out. During tube placement, the mass appeared as a yellow-white bulge that was suspicious for a cholesteatoma. On computed tomography (CT), the mass appeared as a 4 × 2 × 2-mm soft-tissue lesion at the level of the tympanic membrane along the malleus. No osseous erosion was noted.
The patient underwent surgical resection of the lesion followed by a tympanoplasty to reconstruct the defect. Intraoperatively, the lesion was firm, homogeneous, pale yellow, and nonencapsulated (figure 1). It was isolated within the squamous and mucosal layers of the tympanic membrane. The mass was located lateral to the manubrium and did not extend beyond the squamous surface. Posteriorly, it could not be separated from the squamous and mucosal layers. Anteriorly, it was superficial to the fibrous layer and had irregular borders. The middle ear was normal.
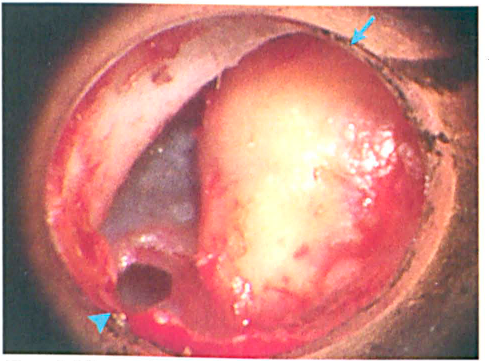
Microscopic view of the left tympanic membrane shows the intratympanic xanthogranuloma (arrow) and a small perforation after removal of the previously placed tympanostomy tube (arrowhead).
Histologic examination of intraoperative frozen sections identified multinucleated giant cells and sheets of histiocytes (figure 2, A). No indications of malignancy were evident. Subsequent immunohistochemical staining was positive for CD68 (figure 2, B) and negative for CDla, which helped clinch the diagnosis of JXG. No skin lesions or café au lait spots were found on physical examination, and the patients family history was negative for any cutaneous skin disorders. The tympanic membrane graft healed well postoperatively, and the patient exhibited no evidence of persistent disease during months of follow-up.
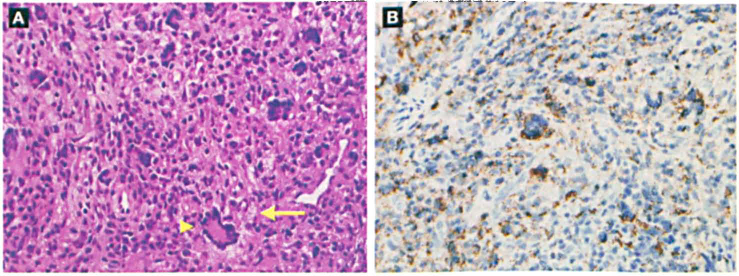
A: Histology reveals a wreath-like Touton giant cell (arrowhead), foamy histiocytes (arrow), and indistinct cellular borders, irregular nuclei, and eosinophilic cytoplasm (H&E, original magnification x400). B: Immunohistochemical staining is strongly positive for the histiocyte marker CD68 (original magnification x400).
Discussion
The tympanic membrane is formed by the fusion of three embryologic layers: the endoderm, the mesoderm, and the ectoderm. The thin epidermal layer contains a dermal layer populated by dendritic cells, including histiocytes. Primary lesions of the tympanic membrane are an unusual diagnosis; among them are intratympanic cholesteatomas, tympanosclerotic lesions, cholesterol granulomas, schwannomas, hemangiomas, squamous cell carcinomas, and meningiomas. Our case adds primary JXG to the list.
JXG is a self-limited disorder that typically begins as one or more red cutaneous nodules that can become yellow, firm, and rubbery. They generally appear in infancy or childhood (mean age at onset: 22 mo); 35% of cases are present at birth, and only about 10% first manifest in adults.3–6 Most lesions spontaneously regress by the time a child reaches the age of 5 years. JXG occurs as a solitary lesion in 80% of cases, and it is systemic in 4% of cases. 7 JXG is 10 times more common in whites than blacks, and there is a slight male preponderance (1.4:1). 6
These lesions most commonly affect the head, neck, and upper trunk, but they can appear on any skin surface. Lesions can also occur in the gastrointestinal tract, kidney, lung, gonads, pericardium, central nervous system, temporal bone, larynx, and eye.8–12
Conditions associated with JXG include neurofibromatosis type 1 (NF-1), Niemann-Pick disease, urticaria pigmentosa, and epilepsy. 6 A thorough physical examination should be performed to identify hepatosplenomegaly, lymphadenopathy, café au lait spots, and other skin lesions. The combination of multiple JXG and NF-1 in a patient is associated with juvenile myelomonocytic leukemia. 6
The etiology and pathophysiology of JXG are not fully understood. There is debate as to whether a reactive or neoplastic process occurs and whether it represents a granulomatous reaction of histiocytes to a physical or infectious stimulus. 6 The cells of origin in JXG are likely dermal dendritic cells. 13 The precursor cells are the CD34+ progenitor cells, which can differentiate into either CD14+/ CD1a- histiocytes (JXG) or CD14-/CD1a+ Langerhans cells (Langerhans cell histiocytosis), depending on cytokine signaling. 9 Thus, JXG and Langerhans cell histiocytosis are both categorized as dendritic cell-related disorders, as opposed to macrophage-related proliferative disorders such as Rosai-Dorfman disease. 13
Thehistologic features of JXG vary through the lifespan of the lesion. New lesions display dense monomorphic histiocytic infiltration of the dermis, with deeper extension in one-third of cases. Older lesions show dense foam cells, Touton giant cells, and fibrosis. The histiocytes demonstrate pleomorphic nuclei with few mitotic figures and an abundant cytoplasm. Touton giant cells are the characteristic finding in JXG, and thus their presence can differentiate JXG from other conditions. 14 Touton cells are large, lipid-laden histiocytes with multiple nuclei arranged around a central island of cytoplasm. 15
Immunohistochemical studies of JXG specimens show positive staining for histiocyte markers, including CD68 and vimentin. JXG lesions also stain positively for factor XIIIa, and HAM-56 and negatively for CD1a, CD3, CD20, S-100 protein, HMB-45, cytokeratins, actin, and desmin. The absence of nuclear grooves and negative staining for S-100 protein differentiates JXG from Langerhans cell disease.
Different forms of JXG have been described based on clinical and histologic features. Subcutaneous lesions, which have been reported to represent about 5% of cases, are typically solitary, deep-seated, and often congenital or perinatal lesions 10 to 20 mm in diameter.7,8 Deep lesions may be more cellular and monotonous and may contain fewer Touton cells. 7 Other forms include micronodular, macronodular, giant, clustered, lichenoid, and plaque-like lesions. 8
Treatment options for JXG are expectant observation, surgical excision, radiotherapy, corticosteroid therapy and, in rare cases, chemotherapy. JXG lesions often regress spontaneously without any sequelae except for atrophic scarring or altered pigmentation. 16 Surgical resection is necessary for symptomatic cases or as an option for diagnostic and cosmetic concerns. Systemic and intralesional injections of corticosteroids have been reported to promote regression of JXG lesions.17,18 Chemotherapy is indicated in rare cases of severe systemic JXG; 15 cases of systemic treatment with corticosteroids and vinca alkaloids, methotrexate, or etoposide have been reported. 18
In conclusion, JXG is a rare, benign, cutaneous lesion of the non-Langerhans cell group of histiocytic disorders. Because it often affects the head and neck, otolaryngologists should be aware of this entity in order to make a correct diagnosis, to formulate a treatment plan, and to understand the possible complications and associated medical conditions. To the best of our knowledge, our case represents the first published report of a JXG of the tympanic membrane.