
Abstract
Trace element enrichment in coals has increasingly been focused in the community of coal geochemistry. To research the enrichment mechanism of trace element in coals on molecular/atomic scale is an important complement of field investigations, but relevant works are scattered. In the present work, the effect of local structure on the ability of main oxygen-containing groups to chelate trace element metal ions including –COO− and C6H5O− is preliminarily quantified. It is completed by evaluating the Mulliken charge excess ξ, i.e. the sum of Mulliken charge for metal ions and atoms involved in chelating functional groups. When electron-donating groups like benzene and its derivatives immediately connect to cation carboxylates, excess electron will be transferred to cation carboxylates, pulling down the ξ values and stabilizing metal–humate complexes. Otherwise, electron-withdrawing groups like C = C, C≡C, and C = O play a contrary role. Based on calculations, at the vicinity of ξ ≈ 0, the binding energy is highest. For monovalent cation ions, the most stable metal–humate complex is that chelated by small alkyl carboxylate, while the highest binding energy is not found for high-valent cation ions in the investigation. Meanwhile, both the combination of hydroxyl and carboxyl and hydroxyls have higher binding energies than the combination of hydroxyl and carbonyl or carboxyls at the same ξ range.
Introduction
In the last decades, enrichment of trace element relative to world average coal has been extensively reported (Dai et al., 2017: Lauer et al., 2017; Long and Luo, 2017; Qin et al., 2015; Saikia et al., 2009, 2015, 2016; Sun et al., 2016; Sun et al., 2017; Xiao et al., 2016; Yi et al., 2015; Zhao et al., 2014, 2017). In some cases, extremely enriched lithium-bearing coal seams are considered as super large lithium deposits if the industrial grade of associated Li2O deposit in pegmatite, 0.2%, is adopted to evaluate the lithium content of coal ash (Sun et al., 2012). It is noted that cell-filling authigenic minerals are relevant to trace element enrichment and are indicative of the ore-forming material from peat accumulation or from the post hydrothermal fluids (Seredin et al., 2013; Sun et al., 2013, 2015). In addition, , trace elements tend to be enriched in the top or bottom of coal seams, even in roof and floor rock samples or the parting samples. However, further investigations of the occurrence of trace elements and their origins in coals have been impeded by the state-of-the-art analytical methods and unambiguous enrichment mechanisms, albeit significant potential availability. Based on field investigations, He et al. (2016) hypothesized that oxygen-containing functional groups on the surface of humic substances, the main organic of low-rank coal, played the role of adsorption site in transporting trace element cation ions, and in subsequent polymerization, trace element cation ions entered into the fluids, then precipitated with authigenic minerals or were exported by fluids. Our previous work has qualitatively revealed that singly deprotonated alkyl/aryl carboxylic acids, analog of humic substance, are able to effectively transport trace element cation ions (He et al., 2016). The less the metal–ligand–bonding part deviates from electroneutrality, the more stable the organometal complex is. Moreover, intensive coalification is shown to be unable to stabilize carboxylates. To decipher these scenarios, the ability to transport and adsorb trace element cation ions by oxygen-containing functional groups on the coal surfaces must be explored as well as the effect of coal structure. Nonetheless, none of the appropriate parameters has been selected to describe what role of the electronic effect of the groups on the charge distribution on singly deprotonated carboxylic acids, i.e. the effect of coal structure on the ability of humate.
In the present work, combining with binding energy (BE) data, population analysis was carried out to quantify the role of electron-donating and electron-withdrawing groups on the stability of the cation carboxylate when immediately connected to this system. In addition to monovalent and divalent cation ions, high field strength elements (HFSEs) such as Sm3+, Eu3+, and Gd3+ were employed to validate our preliminary conclusions. Besides p-benzoquinonyl and carbonyl, more typical electron-withdrawing groups including C=C, C≡C, and C=C-C=C are selected to further probe the effect of electron re-distribution on the stability of Li carboxylate.
Calculation methods
Although the structure of the coal is heterogeneous, its basic structural unit is nearly identical when the rank is the same. With respect to the low-rank coal, its basic structural unit is characterized by one or two benzene ring and oxygen-containing groups on the side chains. To represent the whole coal with basic structural unit, i.e. simple alkyl/aryl carboxylic acids, could effectively simulate the essential part of the coal taking part in all physicochemical reactions and dramatically decreases the computational cost.
Our previous work effectively optimized the geometry of adsorbed metal ions and calculated the BE for monovalent and divalent cations with BP86/Stuttgart Dresden Dunning ECP method (He et al., 2016). Thus, this method is still selected to perform geometry optimization for HFSEs. The BE is calculated by:
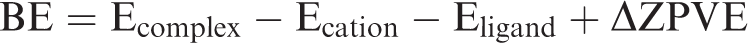
It should be noted that the quantity of electron transferred to/from metal carboxylate is characterized by Mulliken charge excess. Specifically, Mulliken charge excess, represented by ξ, is defined as the sum of Mulliken charge of metal ions and functional groups taking part in the formation of coordination bonds. Usually, ξ values are somewhat less than the total charge of metal–organic complex for electron-donating groups, otherwise higher for electron-withdrawing groups. All geometry optimization, electronic energy, and Mulliken charge calculations were carried out in Gaussian09 software package (Frisch et al., 2013).
Results and discussion
Geometry structures of complexes
Possibly due to the high ionic potential, HFSEs tend to form tridentate with the B site of benzoquinonetetracarboxylic acid where two carboxylates on ortho position chelate HFSE ions at the same time. Also, at the E site of fluvic acid, the deprotonated alcohol hydroxyl with the nearest ether formed bidentate complex with HFSE ions. Except these two ligands, HFSE ions tend to form bidentate complexes like monovalent and divalent metal ions.
Bi-model of BE values plotted with ξ values
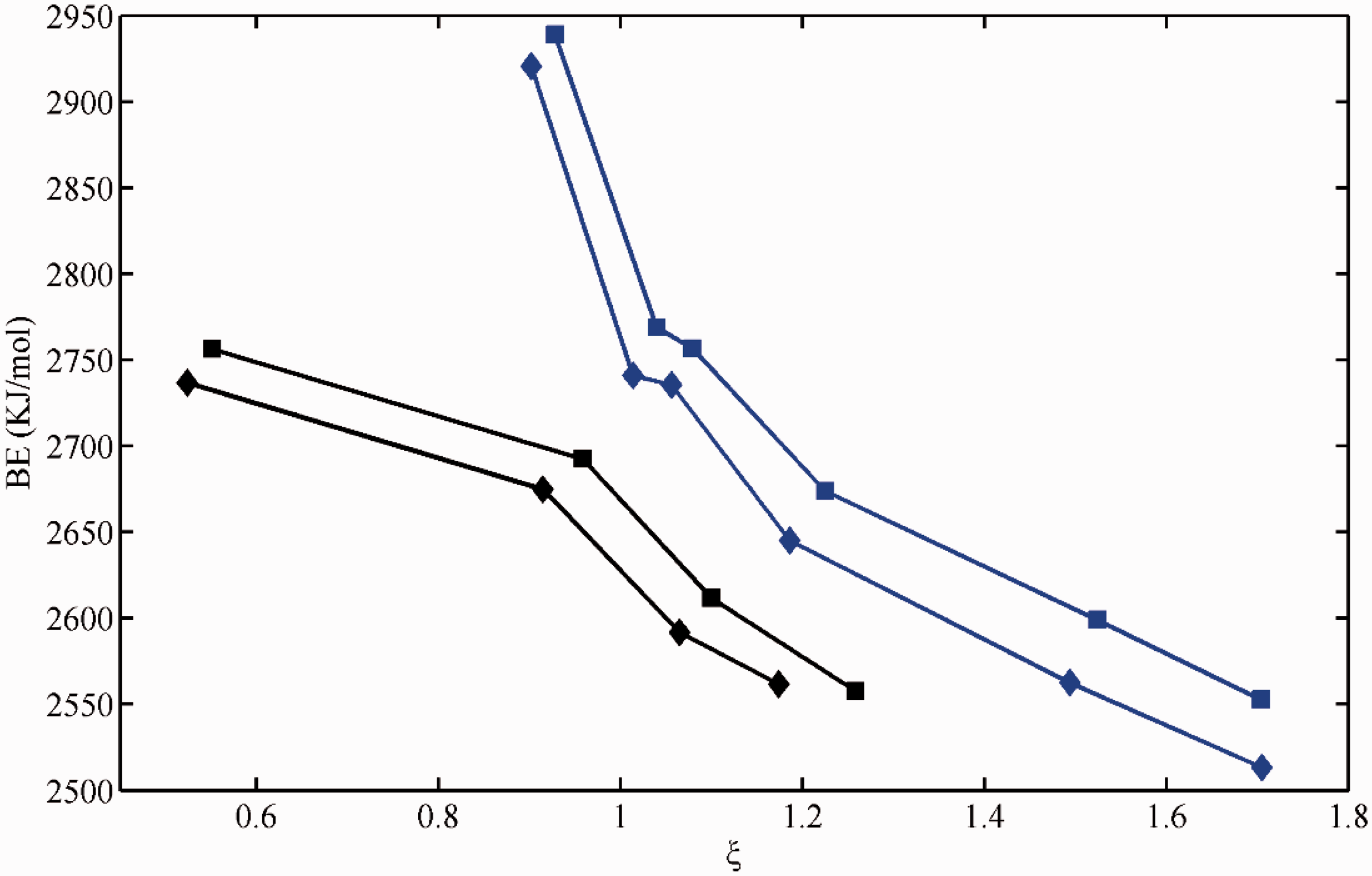
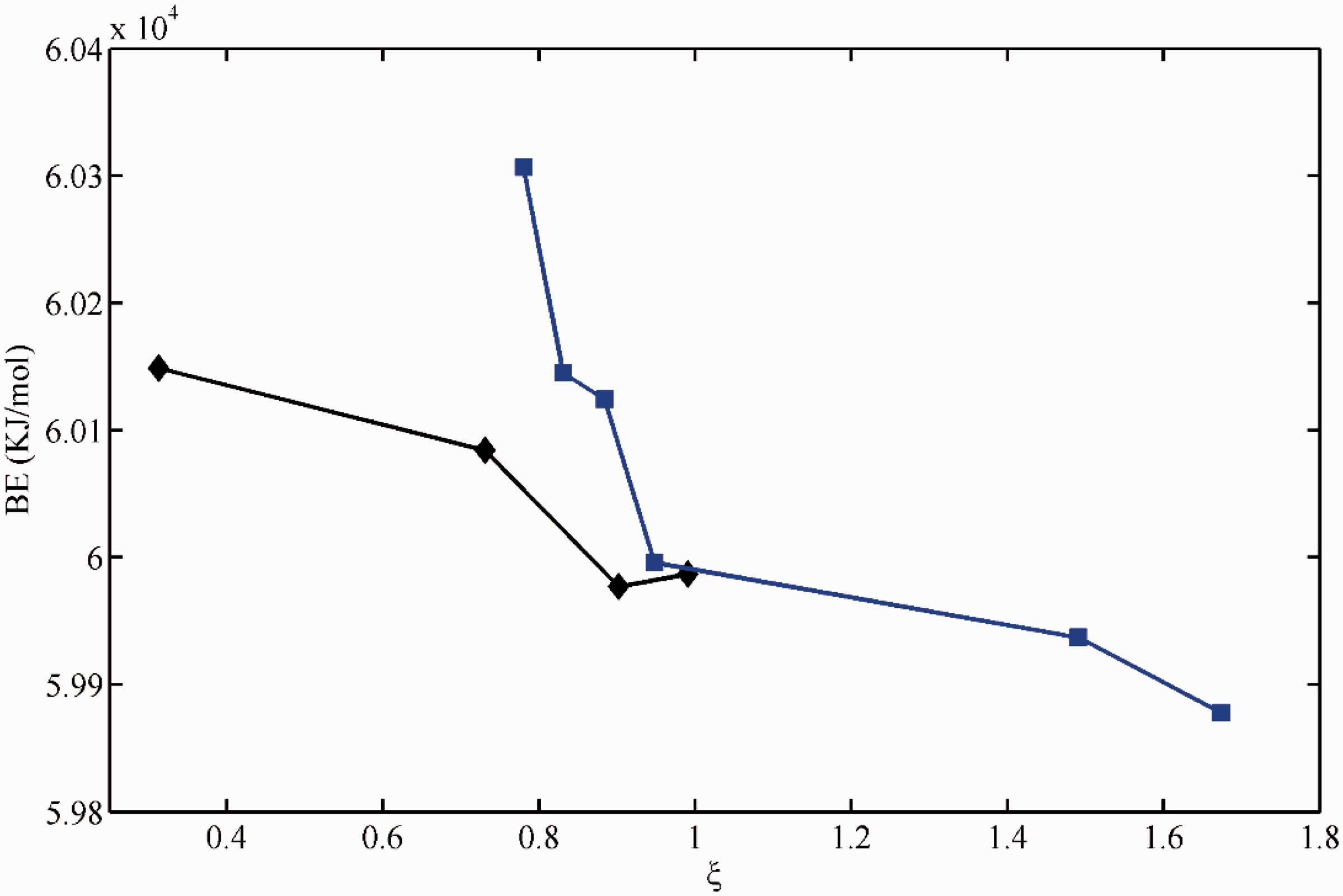
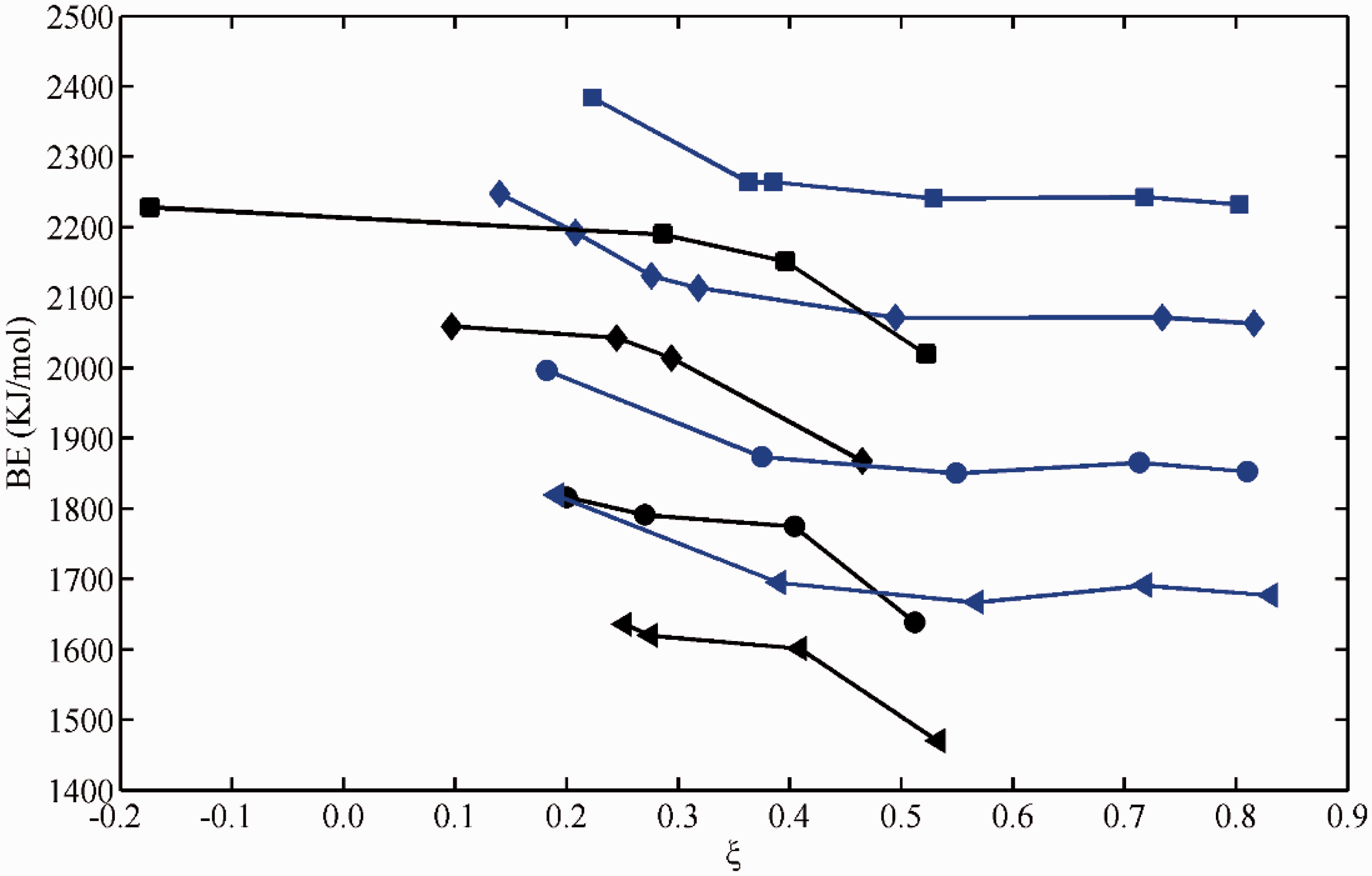
Obviously, for divalent and trivalent cation ions, BE values decrease monotonically with ξ values for both electron-donating groups and electron-withdrawing groups (Figures 1 to 3, Table 1), occurring as bi-model. Meanwhile, the BE values response to electron-withdrawing groups are less than that of electron-donating groups nearly at the same ξ value range. For monovalent cation ions like Li+, an asymmetric parabolic line occurs in the plot of BE values relative to ξ value (Figure 4). The maximum BE value can be found at the vicinity of ξ ≈ 0. Also, the ξ values of typical electron-withdrawing groups including C=C, C≡C, and C=C-C=C are expected to be larger than the total charge of organolithium, i.e. zero.
Variations of binding energy for Sm3+ and Eu3+ against ξ. Data specification: squares for Eu3+ and diamonds for Sm3+; blue lines for electron-donating groups while black lines for electron-withdrawing groups. BE: binding energy.
Variations of binding energy for Gd3+ against ξ. Data specification: blue squares for electron-donating groups while black diamonds for electron-withdrawing groups. BE: binding energy.
Variations of binding energy for Ni2+, Cu2+, Zn2, and Cd2+ against ξ. Data specification: squares for Ni2+, diamonds for Cu2+, circles for Zn2+, and triangles for Cd2+; blue lines for electron-donating groups while black lines for electron-withdrawing groups. BE: binding energy.
Binding energy and ξ values for different carboxylate-containing ligands.
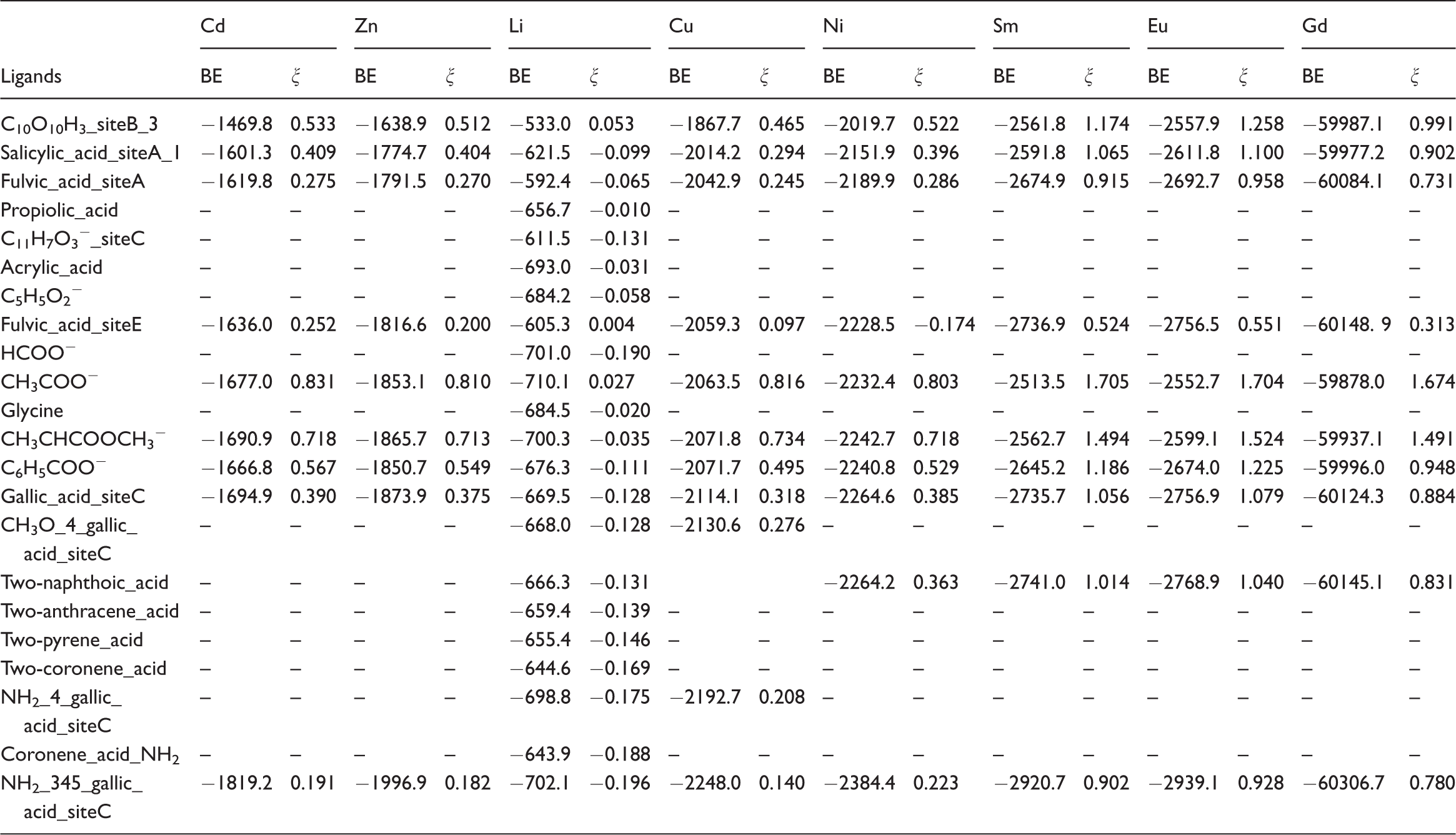
Note: The BE values for Cd, Zn, Li, and Cu cases from He et al. (2016). BE: binding energy.
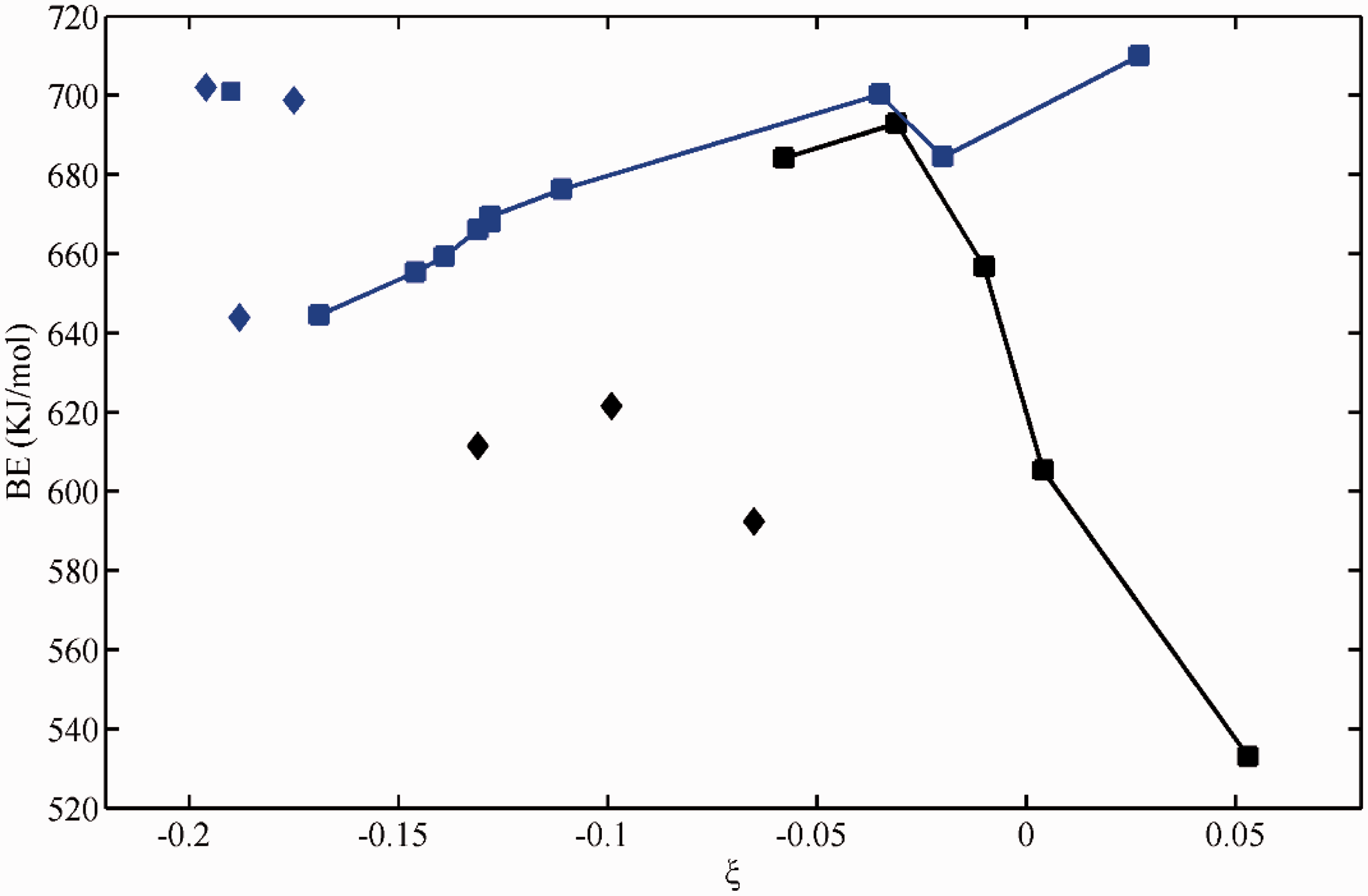
Variations of binding energy for Li+ against ξ. Data specification: blue squares and diamonds for oxygen-containing electron-donating groups without or with amino on the benzene ring, while black squares for electron-withdrawing groups, black diamonds for electron-withdrawing groups generated by steric hindrance. BE: binding energy.
Apparently, A site of sacyclic acid, A site of fluvic acid, and C site of one-hydroxyl-2-naphthoic acid all apparently deviate from the trend line of electron-withdrawing groups (Figure 4). This may attribute to the atypical character of this kind of electron-withdrawing groups generated by steric hindrance in which benzene or naphthalene still acts as electron-donating groups to some extent, evidenced by the negative ξ values. If it is true, the plots of BE with ξ are in good agreement with our previous statement (He et al., 2016). However, there are still four data in discordant with the parabolic line, which correspond to derivatives of gallic acid by amino to partially or totally replace hydroxyls on benzene ring and formate. The ability to transfer electron is high for amino, which generally accounts for the high BE values. Otherwise, in the C site of one-hydroxyl-2-naphthoic acid, the amino group is located on the next benzene ring in which the effect of amino on the BE value is insignificant. Maybe the effectiveness of amino should be distinguished from that of oxygen-containing groups, the data points on the parabolic line. There is no good explanation for the data of lithium formate. At the same time, we gradually elevate the number of aromatic rings conjugated with metal carboxylate to probe the role of coal rank on the stability of complexes. The results showed that the BE value gradually decreased with the decrease in ξ value (Figure 4). It means that the complex will be unstable with the increasing aromaticity of coal basic units for lithium.
At the same time, the influence of oxygen-containing group combination type to the BE of metal–humate complexes is investigated (Table 2 and Figure 5). The BE of the combination of hydroxyls or hydroxyl and carboxyl is higher than the combination of carboxyls or hydroxyl and carbonyl. This may be the result of unsaturated ring connected to oxygen-containing groups in the latter.
Binding energy and ξ values for different combinations of oxygen-containing functional groups.
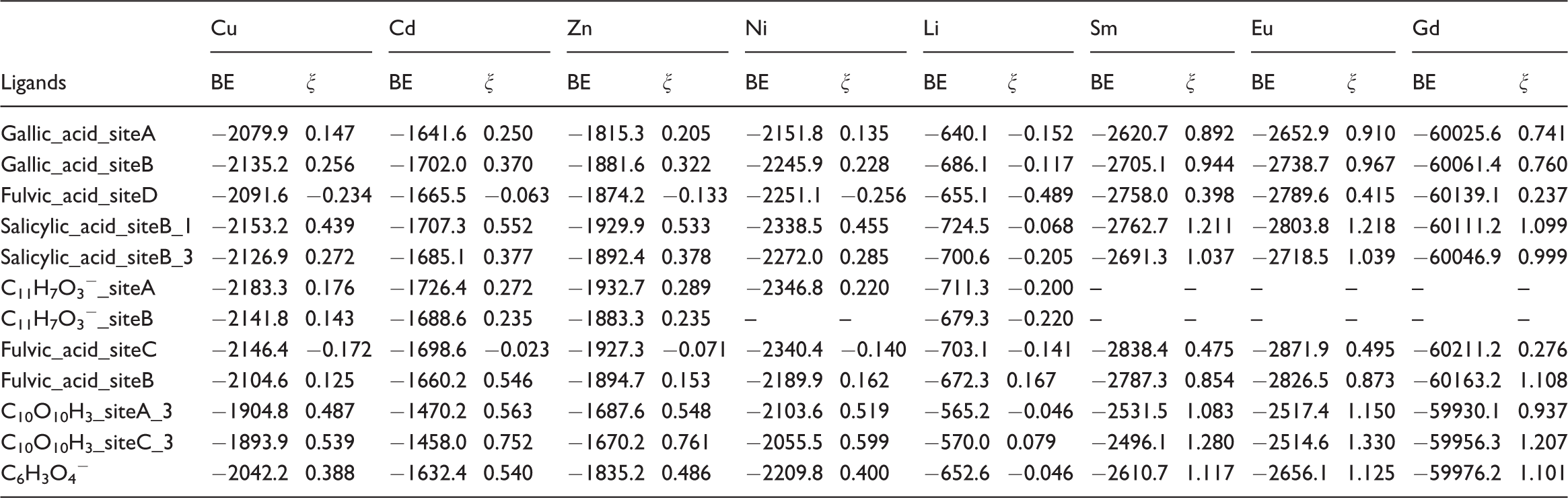
Note: The BE values for Cd, Zn, Li, and Cu cases from He et al. (2016). BE: binding energy.
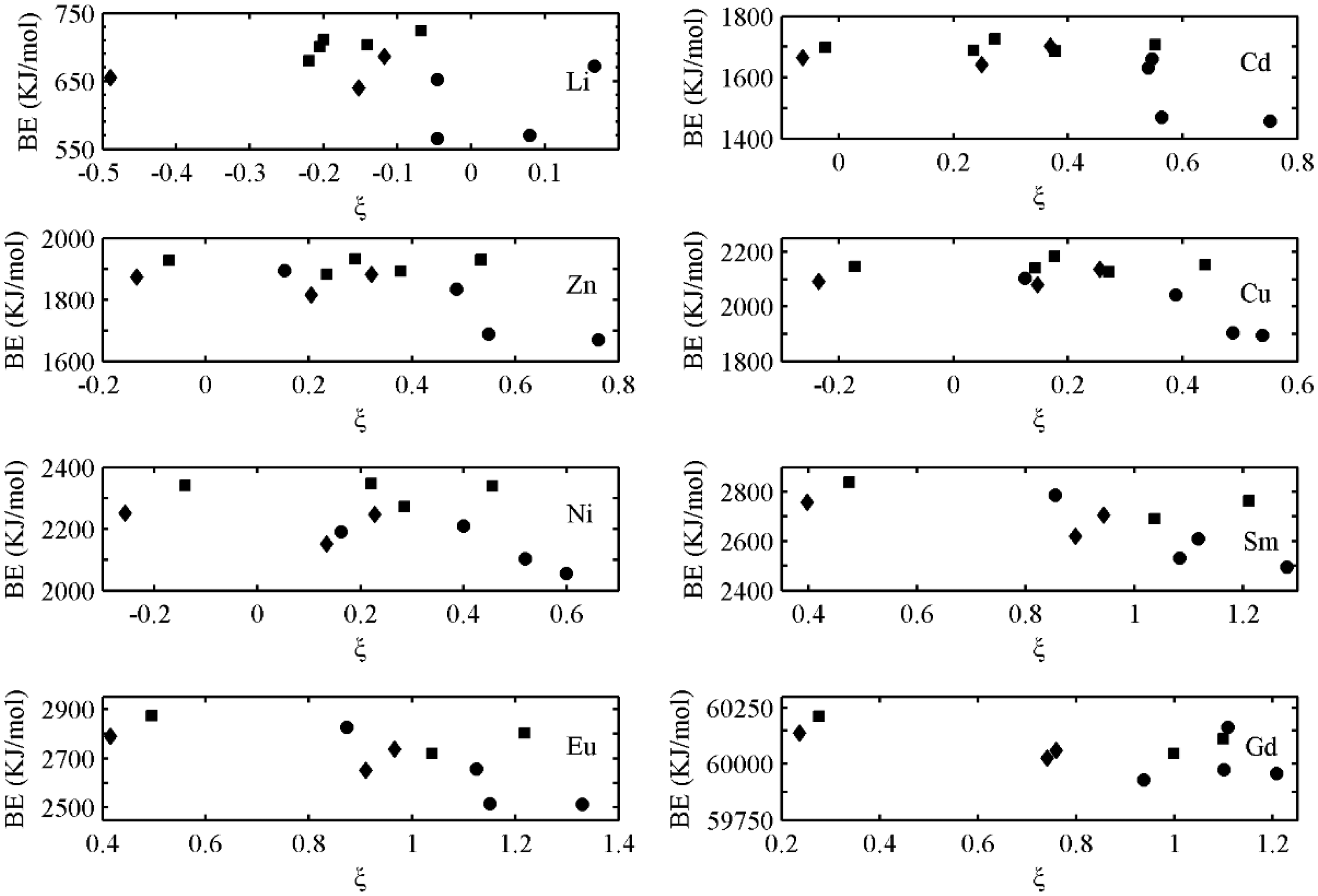
Binding energy of different combinations of oxygen-containing functional groups plotted against Mulliken charge excess, ξ. Symbol notation: squares: hydroxyls; diamonds: hydroxyl and carboxyl; and circles: carboxyls or hydroxyl and carbonyl. BE: binding energy.
Implications to the potential of trace element to be enriched in coalification
For monovalent cation ions, small alkyl carboxylate anions are capable of effectively chelating them. However, alkyl carboxyl dominates the moity of carboxyl in original materials of sapropelite. Therefore, does it point to the direction of exploring lithium enriched coal in sapropelite? More possibly, small alkyl carboxyl occurs as the irregular part of the basic unit of the coal. In coalification, the content of lithium in coals drops dramatically with the decrease in small alkyl carboxyl content. Although the ability to transfer electron is large for amino groups, the predominant species of nitrogen in coals are pyrrole and pyridine which do not belong to strong electron-donating groups.
For high-valent cation ions, the occurrence of larger conjugation systems in coalification is expected to facilitate the enrichment of trace element, but the decrease in oxygen-containing groups will suppress this process to a large extent. Thus, a maximum content of trace element may be found in a certain stage of coalification.
Furthermore, as to the strong affinity of Gd3+ to oxygen-containing groups apparently distinguishing from that of Sm3+ and Eu3+, whether LREE3+, MREE3+, or HREE3+ could show different geochemical behaviors when interacting with organic matters? Besides, higher valent trace elements including Se, Cr, Nb, and Ta usually exist as anions in Earth surface environments and likely have different geochemical characteristics from cation ions. As to now, there are not enough published works to discuss the genesis of trace element enriched coals. To clarify these problems necessitates more works on molecular/atomic scale.
Conclusions
In the present work, the effect of local structure on the ability of main oxygen-containing groups including –COO− and C6H5O− is preliminarily quantified. It is completed by evaluating the Mulliken charge excess ξ, i.e. the sum of Mulliken charge for metal ions and atoms involved in chelating functional groups. When electron-donating groups like benzene and its derivatives immediately connect to cation carboxylates, excess electron will be transferred to cation carboxylates, pulling down the ξ values and stabilizing metal–humate complexes. Otherwise, electron-withdrawing groups like C=C, C≡C, and C=O play a contrary role. Based on calculations, at the vicinity of ξ ≈ 0, the BE is highest. For monovalent cation ions, the most stable metal–humate complex is that chelated by small alkyl carboxylate, while the highest BE is not found for high-valent cation ions. Meanwhile, the combination of hydroxyl and carboxyl and hydroxyls has higher BEs than the combination of hydroxyl and carbonyl and carboxyls at the same ξ range.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: State Natural Sciences Foundation Youth Program (No. 41603011), Hebei University of Engineering doctoral special program (No. 17129033019 and No. 17129033020), and Natural Science Foundation of Hebei Province (D2016402104).