
Abstract
Binding characteristics of metal ions (Li+, Cd2+, Zn2+, Cu2+) with humic substances (HSs) are investigated by first principle calculation at BP86/SDD level. In the present work, HSs are well represented by singly deprotonated alkyl/aryl carboxylic acid. The results reveal that the bidentate is the strongest complexation form. Moreover, with the increase of ionic potential, the binding energies of HSs to chelate metal ions increase in order, Li+ < Cd2+ < Zn2+ < Cu2+. The nature of the ligand also exerts important influence on the stability of organometallic complexes. Specifically, for Li+, the ionic bond-forming part occurs as electroneutrality. Whatever electron-donating groups or electron-withdrawing groups including alkyl, aryl and its derivatives (salicylic acid and m (p)-substituted gallic acid by hydroxyl or amino) and carbon-carbon double bond connects to the carboxylate group, the electron density of the ionic bond-forming part will be changed resulting in the decrease of binding energy (BE). However, for ions with high ionic potential or high positive charge density (Cd2+, Cu2+ and Zn2+), the ionic bond-forming part is positively charged. From electron-withdrawing groups to electron-donating groups, the extent to deviate from electroneutrality is gradually decreased, although the quantity of electron is possibly insufficient to balance the extra positive charge. As a result, the BE value gradually increases, but does not reach a maximum for studied ions and ligands. Besides, mixed functional groups are able to stabilize organometallic complexes. The optimal mixed functional group is carboxyl and hydroxyl on ortho position, followed by two hydroxyls, hydroxyl and carbonyl, carboxyl and carbonyl, two carboxyls. Strong electron-withdrawing effect attributes to the decrease of the BE value for the latter three.
In the end, the geochemical behavior of trace elements in coalification is predicted: small organic acids generated by decomposition of dead plants in relatively oxidized environments will facilitate the enrichment of lithium and this proportion of lithium will be released into pore water in the polymerization of small molecules, possibly accumulating in secondary minerals in low-rank coals; metal ions with high ionic potential bonded by organic molecules will be released in a later stage and accumulate in slightly higher rank coals.
Introduction
Recently, a variety of trace elements including Li, Ge, U, Hg is reported to be enriched in coal seams worldwide (Dai et al., 2014; Qin et al., 2015a, 2015b; Seredin et al., 2013; Sun, 2015; Sun et al., 2012, 2013a, 2013b, 2016). Most of these studies focused on the anomalous concentration of trace elements in coals and the affinity of these elements to the organic matters or the inorganic parts can be discerned on the level of mineral (Chu et al., 2015; Kang et al., 2014). However, the studies on the enrichment mechanisms and the origin of these elements in coals are scared (Qin et al., 2015b) and badly needed. Ambiguous recognition to these key problems brings about the low efficiency of coal utilization and extreme exacerbation of the environment due to the release of hazardous elements. To probe the modes of occurrence of trace elements in details, i.e. the chemical bonding environments around the trace element atom of interest will shed light on the enrichment mechanism and provide suggestions to coal clean utilization.
To deduce the enrichment process is a difficult work since the coalification is sluggish and the structure of coal is complex. From this perspective, except vibrational spectroscopy, ab initio or first principle calculation is the best selection to assist unraveling this scenario. First principle calculations are supposed to provide exact electronic structure calculation and molecular dynamic simulation.
Besides the way to be brought by hydrothermal fluids, valuable/hazardous elements can also be enriched in peat or low-rank coals at (syn-) sedimentary stage (Sun et al., 2010; Wang et al., 2015). The structure of the peat or the low-rank coal can be represented by humic substances (HSs). This facilitates the simulation of element enrichment in coals since candidates of HSs structure are intensively researched in previous studies (Gao et al., 2015; Ramalho et al., 2007; Sviatenko et al., 2016; Zhu et al., 2015). Although the concrete molecular structure of HSs has yet not been determined due to its large molecular size and complexity, the main functional groups able to form complex with metals include carboxyl, hydroxyl, and phenol of local configuration. Sundararajan et al. (2011) showed that the predominant of adsorptive site for uranyl ion was the carboxyl and the functional groups nearby might aid the chelation. For other metal ions with smaller size, whether identical adsorption behaviors can be observed has yet not been intensively investigated. In the present study, first principle calculations are employed to investigate the characteristics of Li+, Cu2+, Cd2+, and Zn2+ bonded to HSs, mimicking the enrichment of element in coals. Specifically, we focus on the ability of all favorable functional groups to chelate metal ions in terms of binding energy (BE) and discuss the factors influencing the stability of complexes.
Calculation methods
The symmetric νs(C=O) strength mode calculated at different levels.

Experimental data comes from Ibrahim (2009).
From Table 1, the symmetric νs(C=O) strength mode calculated at BP86/SDD level is in good agreement with the experimental data. At the same time, we have compared the optimized geometry (Cd(H2O)62+ at BP86/SDD level) with EXAFS data (2.27 Å vs. 2.30 Å, Vasconcelos et al., 2008). In the end, this level has been selected to optimize geometry and calculate BE since its good performance and rather lower computational cost (not shown).
Geometry optimizations are carried out for all candidates of HSs, including multiple adsorption sites on one candidate (Figure 1). Additionally, harmonic vibrational frequencies were analyzed to ensure that the geometries obtained represent local minima, to say the least, on the potential surface. All calculations are implemented with the GAUSSIAN09 software package (Frisc et al., 2010). Computational resources for the present work are provided by the National Supercomputer Center in Guangzhou (NSCC-GZ).
Multiple adsorption sites on HSs candidates involved.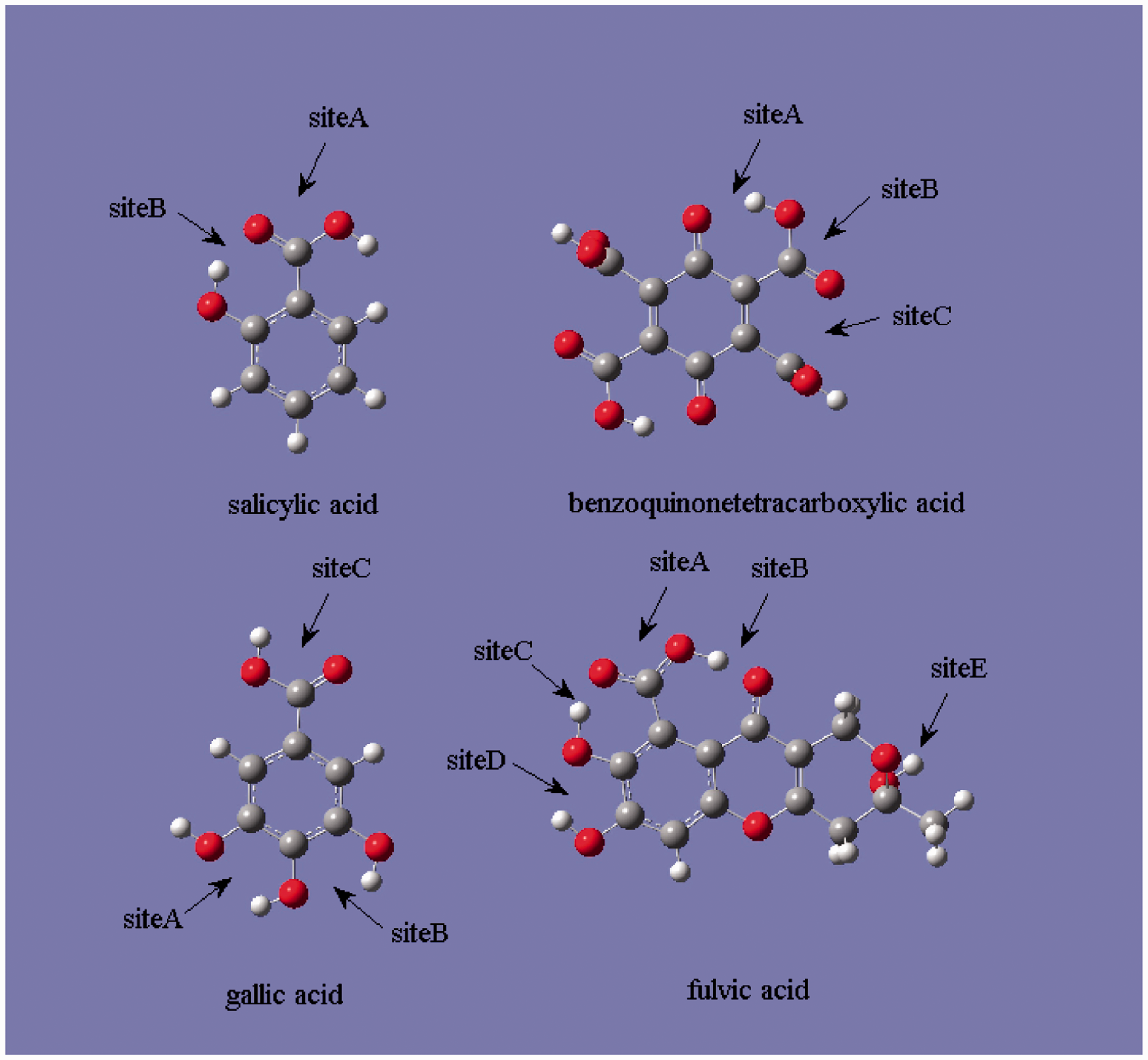
Herein, the binding energies of functional groups to chelate Cd2+, Zn2+, Cu2+, and Li+ are calculated with BP86/SDD level, in which all atoms are described by Stuttgart/Dresden ECP. The specific calculation of BE follows:
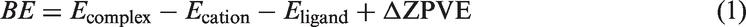
Results and discussion
Geometry of adsorbed metal ions
The most stable metal-ligand complexes occur as bidentates shown in Figure 2 (similar geometries of Cd2+, Zn2+ and Cu2+ are not shown herein). It can be seen that except the carboxylate anion, ortho-substituted oxygen-containing groups on benzene or heterocyclic organics are either able to stably chelate metal ions. Specifically, in the case of carboxylate, both the oxygen atom from deprotonated hydroxyl and the oxygen from carbonyl all take part in binding with Li+. Whilst in other cases, part of carboxyl participates chelation together with neighbor functional groups including hydroxyl, the other carboxyl or quinone groups. Besides, two hydroxyl groups can also complete the chelation.
The binding types of ions to different candidates of HSs.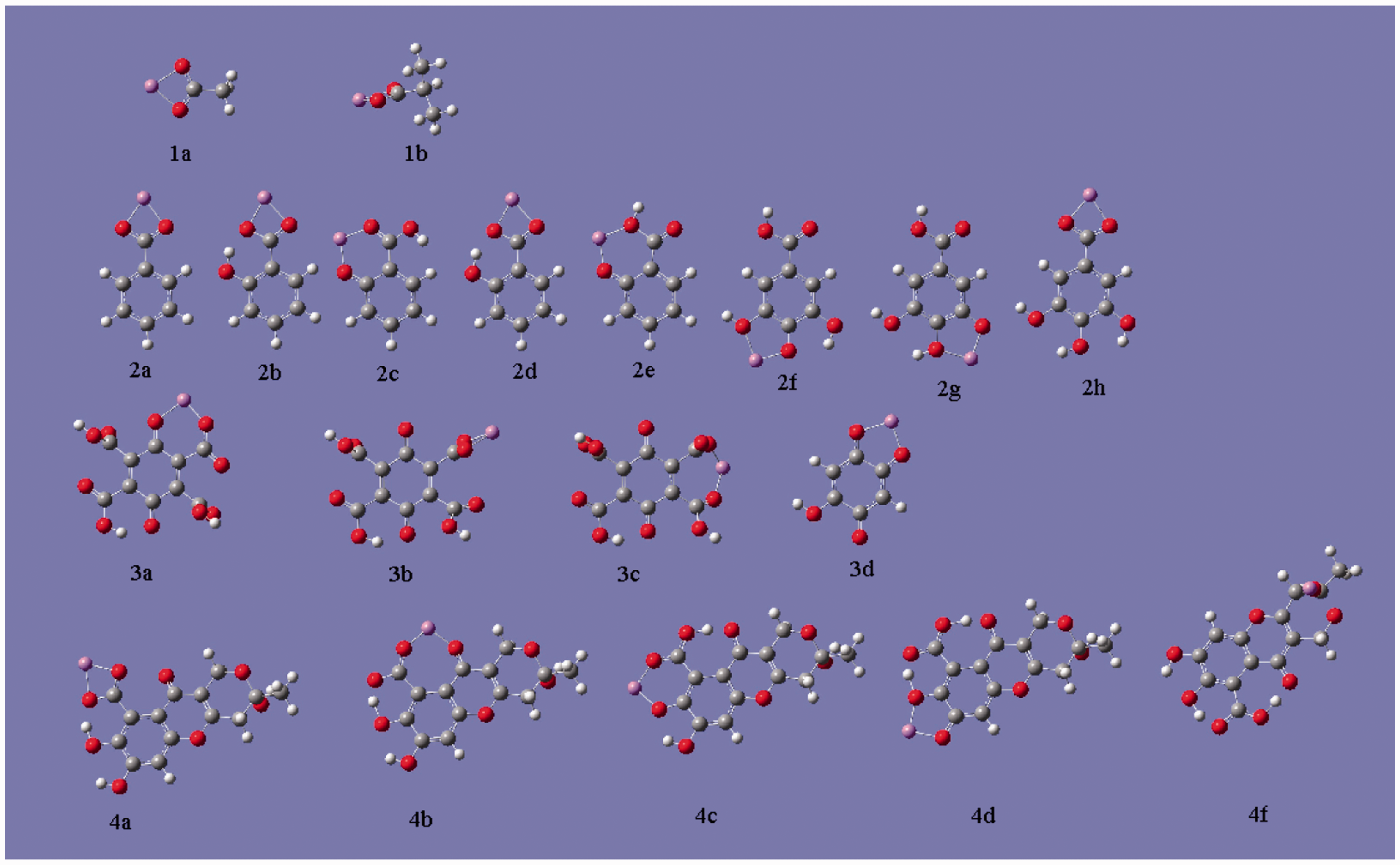
The role of ionic potential of cations
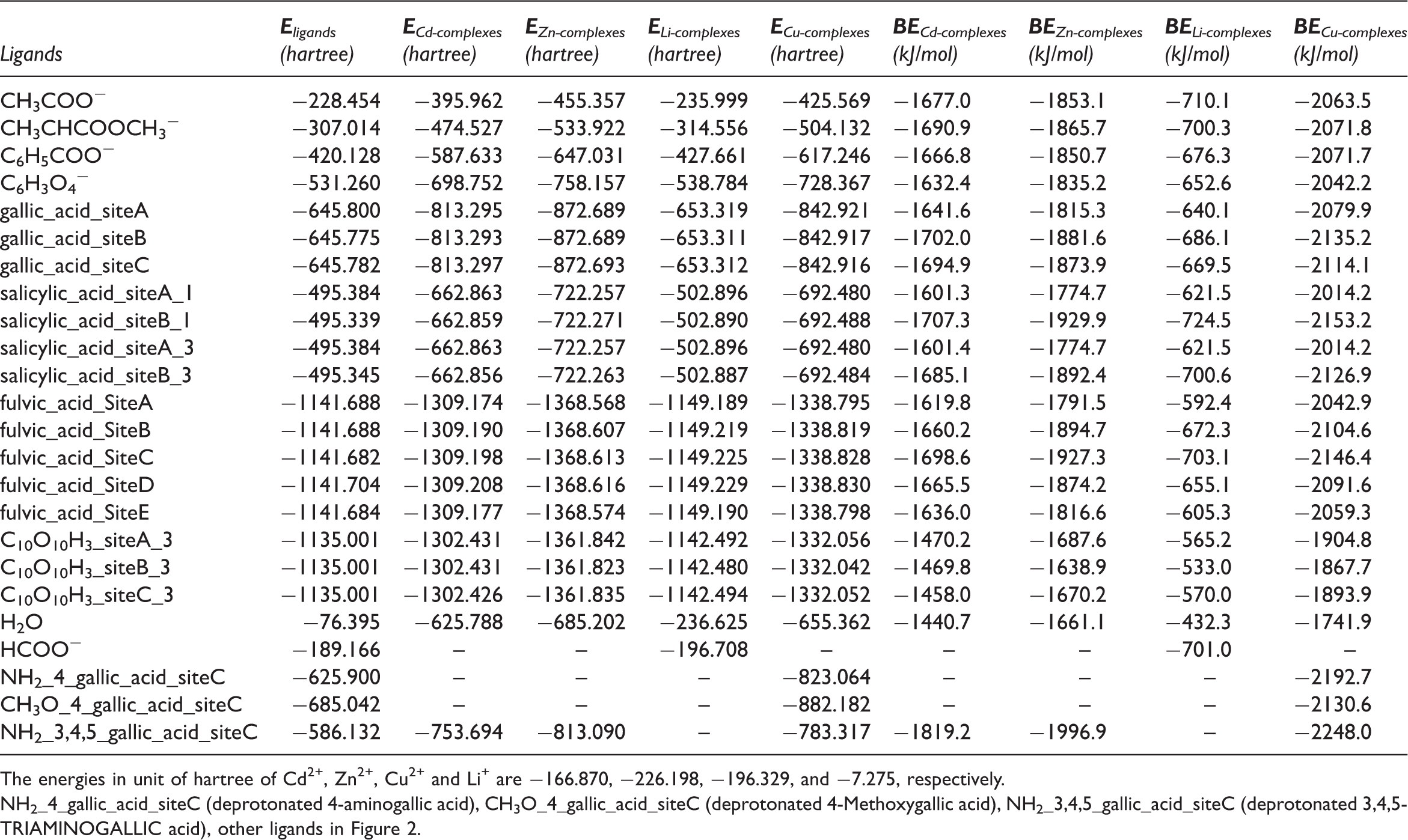
As is shown in Figure 3 and Table 2, with the increase of ionic potential, the binding energies of all functional groups with metals increase from ca. 600 kJ/mol to 2000 kJ/mol, showing the identical trend for four metal ions. Although Cd2+, Zn2+, and Cu2+ are separated into borderline ion category, which seek for nitrogen/sulfur in organic matters, while Li+ referred as hard ion category seeking for oxygen, they can form more stable complexes when reacting with oxygen-containing functional groups relative to hard ions (Nieboer and Richardson, 1980). This statement greatly agrees with our results.
The binding energy of complexes variation with the ionic potential of cations. Binding energies of all complexes. The energies in unit of hartree of Cd2+, Zn2+, Cu2+ and Li+ are −166.870, −226.198, −196.329, and −7.275, respectively. NH2_4_gallic_acid_siteC (deprotonated 4-aminogallic acid), CH3O_4_gallic_acid_siteC (deprotonated 4-Methoxygallic acid), NH2_3,4,5_gallic_acid_siteC (deprotonated 3,4,5-TRIAMINOGALLIC acid), other ligands in Figure 2.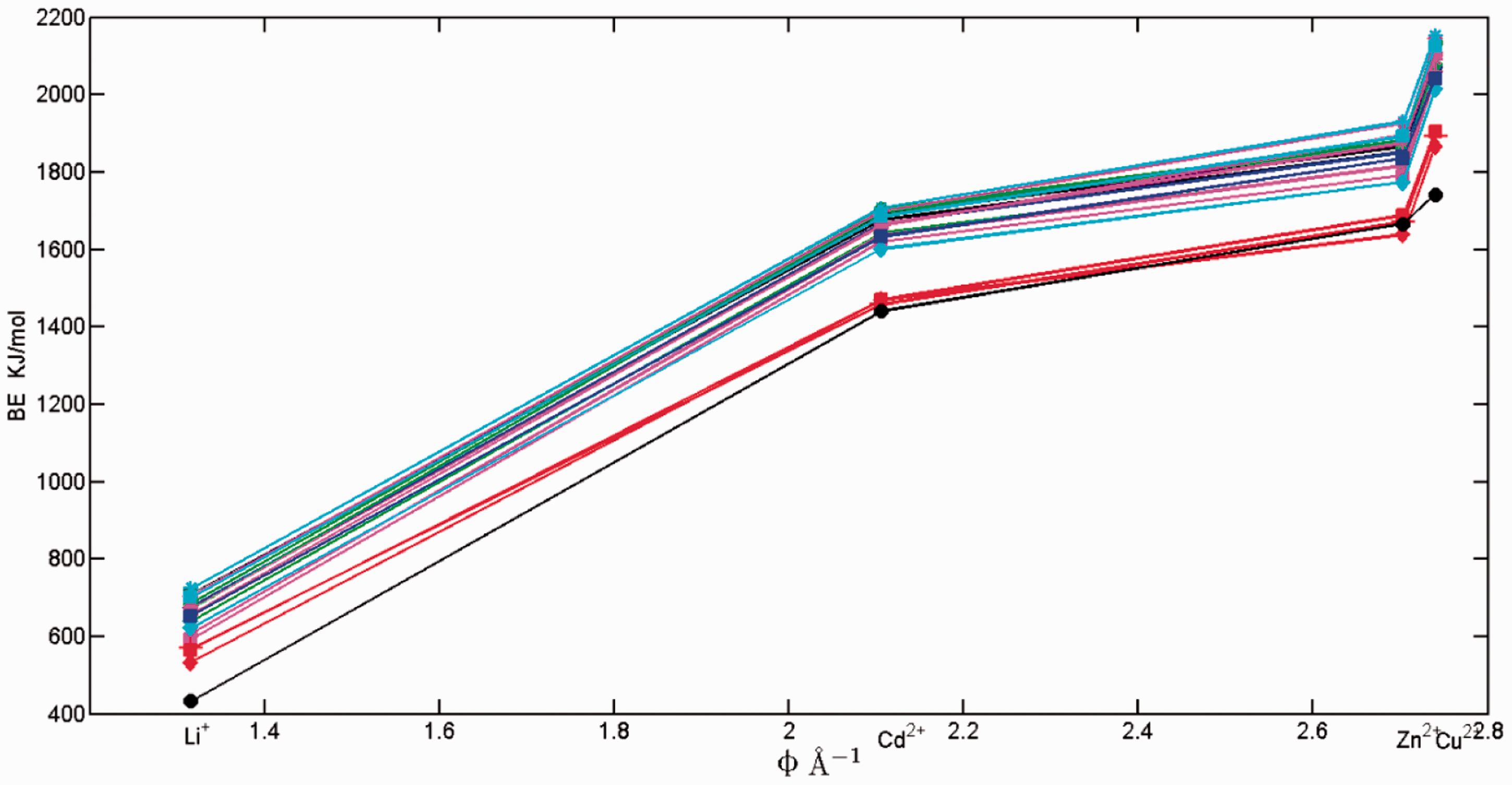
The role of ligand
The nature of the ligand, specific functional group, and the combination of multiple functional groups on local configurations of HSs determine the ability of ligand to chelate metal ions.
The binding energies for the single functional group of the ligands to chelate metal ions are depicted in Figure 4. In general, the carboxyl is considered as the main adsorption site. From Figure 3 and Figure 4, one can learn that due to the limitation of electron transferring/sharing, hydrated metal ions almost have the lowest BE values. Admittedly, the negative charge generated by the deprotonation of carboxyl needs to be delocated, i.e. sharing by carbon and oxygen in the conjugated system, which is necessary for the stabilization of carboxylate anion. We speculate the more charge remains on carboxylate anion whatever positive or negative, the less stable the complex is. In the case of Li+, the ionic bond-forming part occurs as electroneutrality. When strong electron-withdrawing group like carbon–carbon double bond connects to the carboxylate, the electron will be strongly withdrawn from the carboxylate anion resulting in the deviation from electroneutrality for the ionic bond-forming part. This largely reduces the stability of complex. Due to the fact that ortho hydroxyl forms hydrogen bond with benzoic acid and develops steric hindrance, the destruction of conjugation between the aryl and the carboxylate changes benzene ring of the salicylic acid from week electron-donating group to electron-withdrawing group. It also deviates the electroneutrality, then decreases the BE value of organolithium complex, although the extent is lower than that corresponding to deprotonated benzoquinonetetracarboxylic acid. Moreover, when Li+ is chelated by functional groups being less influenced by apparent electronic effect (electron-donating effect or electron-withdrawing effect) like alcohol and formic acid, the BE value increases for the further less deviation from electroneutrality. When methyl or isopropyl connects to the carboxylate, the ionic bond-forming part gains more electrons since the alkyl is electron-donating group. The more the methyl directly/indirectly connects to the central carbon of the carboxylate, the more electrons are obtained. This arrangement will convert the ionic bond-forming part from electroneutrality to electronegativity decreasing the stability of organolithium complex. Comparing to the alkyl, the aryl and its derivatives (m (p)-substituted gallic acid by hydroxyl or amino) conjugated with the carboxylate can offer even more electrons since electrons belonging to big π and p-π conjugated systems can be delocated into the carboxylate and elevate its electron density. It is clear that for organolithium complexes, deviations from electroneutrality on ionic bond-forming part all decrease the BE values (Figure 4). Therefore, the maximum of BE value occurs at the vicinity of lithium acetate.
The binding energies corresponding to a single functional group on the ligand to chelate Li+ (a), Cd2+ (b), Zn2+ (c), and Cu2+(d).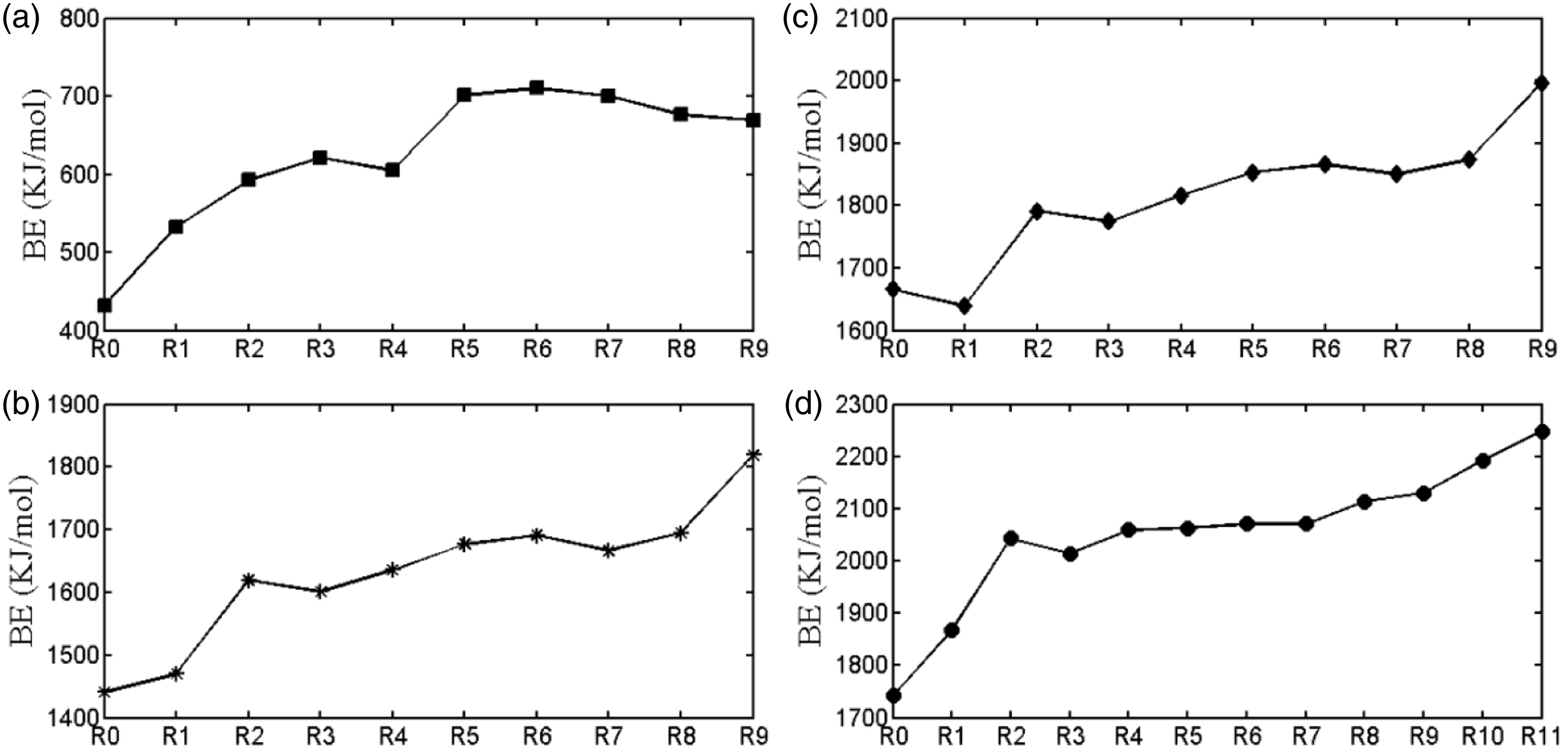
However, for borderline ions, due to the high ionic potential or positive charge density, the ionic bond-forming part is positively charged. From benzoquinonetetracarboxylic acid to 3,4,5-triaminogallic acid, the quantity of electron obtained by the carboxylate anion gradually increases, with the synchronous decrease of the extent to deviate from electroneutrality. This process will increase the BE value. That the BE values for Cd2+, Zn2+ and Cu2+ do not reach the maxima in our study possibly attributes to the insufficient electron gained by the carboxylate to balance the positive charge. It is not unconceivable that the BE value hardly reaches the maximum especially for high strength field element like REE3+. From the perspective of element enrichment and heavy metal removal, the quantity of low-rank coal seems to be the sole factor considered for ions with high ionic potential.
The strongest combination of mixed functional groups to chelate metal ions is hydroxyl and carboxyl (supported by Sundararajan et al., 2011), followed by two hydroxyls, hydroxyl and carbonyl, carboxyl and carbonyl, two carboxyls in the decreasing order (Figure 5). As to this sequence, the latter three are largely influenced by electron-withdrawing groups, i.e. carbon–carbon double bond or carbon–oxygen double bond in the candidates selected. On the other hand, the decrease of BE values seemly correlates with the combinations, albeit the absence of a proper quantity needed to describe the x dimension. In future, more detailed works are needed to probe the ability of mixed functional groups in distinct chemical environments to chelate metal ions.
The binding energies of mixed functional groups to chelate metals, Li+(a), Cd2+(b), Zn2+(c), and Cu2+(d). BE, binding energy; R1, hydroxyl; R2, carboxyl; R3, carbonyl.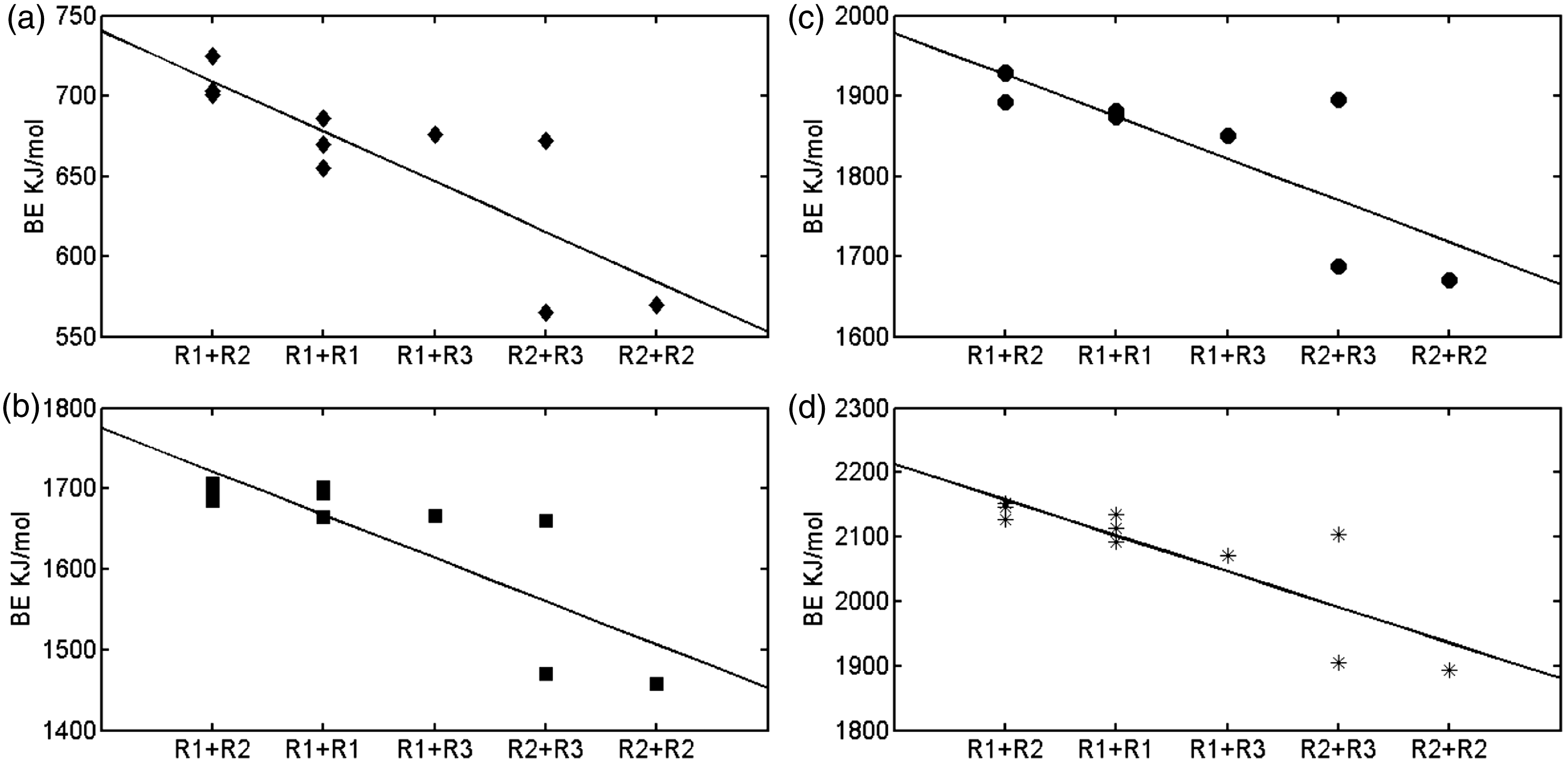
Implication to the enrichment of elements in coals
In our calculations, HSs show considerable potential to chelate metal ions, which maybe an important process to accumulate valuable/hazardous element in peats. Coincident with gradual coalification, simple organic molecules condense into large molecule and oxygen-containing functional groups on side chains are cut in further compaction and metamorphism. In this process, the number of hydroxyl and carboxyl rapidly diminishes and carboxyl is nearly lost in typical bituminous coals. The diminishment of these main functional groups able to chelate metal ions exerts an important influence on the variation of trace element in the organic fraction, which maybe assist the conversion between organic affinity and inorganic affinity.
In relatively oxidized environments, the dead plants are decomposed into small organic acid which facilitates the enrichment of lithium. When small molecules are polymerized, Li+ delivered by small molecules will be released into pore water with a considerable amount. This proportion of ions may enter into the epigenic minerals with low solubility or the leaching fluids off the coal seams. Lithium is expected to be enriched in low-rank coals. However, for metal ions with high ionic potential like Cd2+, Cu2+, Zn2+, even REE3+, these elements will be largely enriched in weekly oxidized environments where relatively large organic molecules containing conjugated electron-donating groups like benzene ring and its derivatives are predominant. These elements will be possibly accumulated in slightly higher rank coals.
In fact, high concentrations of trace element in coals are supposed to be ascribed to elevated content of secondary minerals therein, which is obvious for the vitrinite and the inertinite (Sun et al., 2013a). Trace element bounded to the organic fraction only contributes to a little proportion of the total amount. Based on our calculations, HSs or macerals indeed serve as a transporter. Chelation by simple organic acids and re-release during the formation of macromolecules promote to the transportation and subsequent local accumulation of trace elements, for example in plant cells. However, details involved have yet not been unambiguously understood. Eventually, the geochemical behaviors of trace elements in coalification are complex. More researches are needed to resolve these puzzles.
Conclusions
In the present study, we optimized organometallic complex geometries and calculate binding energies of HSs to chelate metal ions (Li+, Cd2+, Zn2+, Cu2+). Main findings are listed here:
The most stable organometallic complexes are bidentates. With the increase of ionic potential, the binding energies of HSs to chelate metal ions increase in order, Li+ < Cd2+ < Zn2+ < Cu2+. In response to the decrease of the extent to deviate from electroneutrality for the ionic bond-forming part, an obvious increase of BE is found for Cd2+, Cu2+ and Zn2+ and the maximum does not appear in our study. In the case of Li+, the maximum of BE occurs at the vicinity of lithium acetate. The intensity of influence from mixed functional groups on the stability of complex follows the order: carboxyl + hydroxyl > two hydroxyls > hydroxyl + carbonyl > carboxyl + carbonyl > two carboxyls. Strong electron-withdrawing effect attributes to the decrease of the BE value for the latter three. We predict the geochemical behavior of trace elements in coalification: small organic acids generated by decomposition of dead plants in relatively oxidized environments will facilitate the enrichment of lithium and this proportion of lithium will be released into pore water in the polymerization of small molecules, possibly accumulating in secondary minerals in low-rank coals; metal ions with high ionic potential bonded by organic molecules will be released in a later stage and accumulate in slightly higher rank coals.
Footnotes
Acknowledgment
The author thank Dr Zhao Cunliang for detailed discussion.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: State Natural Sciences Foundation Monumental Projects (No. 41490635) and key program (No. 41330317).