
Abstract
A new method for exploiting natural gas from methane hydrates, which combines carbon dioxide replacement and depressurization, was investigated in a 288-ml high-pressure vessel of 288 ml. Experimental results with and without depressurization were compared to reveal the effects of depressurization on the final replacement percentage. Results showed that the CH4–CO2 replacement reaction could be divided into two stages. The first stage lasts for about 4 h and can be described as the surface reaction stage; the reaction in this stage is rapid, and the results fit the Avrami model. In the second stage, the rate of the reaction is slow due to hydrates blocking the diffusion pathways. As the replacement reaction is restricted by the diffusion and transportation of CO2, partial melting of methane hydrates through depressurization enhances the second stage. This is because the melting of methane hydrates provides pathways for CO2 to penetrate into the inner layer of the hydrates. The combined method improves the replacement percentage. However, the effects of depressurization on the stability of the sediments remain to be explored.
Introduction
Natural gas hydrates are crystalline solids formed by guest molecules trapped in water cavities (Sloan and Koh, 2007). They exist below the ocean floor and in permafrost (Chen et al., 2007; Ota et al., 2005; Salehi et al., 2014; Wang, 2010). In the process of exploiting natural gas from methane hydrates, researchers have explored four ways to decompose the crystalline inclusion compounds: depressurization, thermal stimulation, inhibitor injection, and carbon dioxide replacement (Lee et al., 2010). Among these methods, CO2 replacement offers stable long-term storage of a greenhouse gas, while benefiting from CH4 production without heat requirements (Ersland et al., 2009). It also stabilizes the ocean floor (Park et al., 2006) without causing geological deformation. Furthermore, CO2 hydrates are thermodynamically more stable than CH4 hydrates (Ersland et al., 2009). As a result, this concept has attracted much attention.
The concept of injecting CO2 into the CH4 hydrate layer, under the seafloor and permafrost, to replace CH4 gas was first proposed by Ebinuma (1993). As a key indicator of commercial exploitation of CH4 hydrates by replacement, replacement percentage has been the focus of research. (Lee et al., 2003) estimated the limiting composition of mixed hydrates in the bulk phase based on nuclear magnetic resonance spectra data. They found that at least 64% of CH4 could be recovered due to the considerable enrichment of CO2 in hydrates with respect to the gas. Meanwhile, McGrail et al. (2004) assumed that the mole fraction of CH4 would be reduced to approximately 0.48 in the hydrate and increased to a value of 0.7 in the gas phase at equilibrium when using CO2 gas. This was because of the differences in chemical affinity for CO2 versus CH4 in the structure I (sI) hydrate. While experimentally the replacement percentage was below the idealized value predicted through simulation, Ota et al. (2005) conducted experiments with liquid CO2.The replacement percentage for CH4 in hydrates was about 35% after a 307-h replacement on a bulk scale using in-situ Raman spectroscopy. Zhou et al. (2008b) reported that 13.2% CH4 was replaced after 43 h. The reaction conditions were below the CH4 hydrate equilibrium line but above the CO2 hydrate line. Through decades of studies on enhancing the efficiency of the replacement reaction, the state of CO2 used to replace CH4 has been replaced from gaseous CO2 to liquid CO2 and finally to CO2 emulsion (Goel, 2006; Gudmundsson et al., 1997; Zhou et al., 2008b).
Recently, Zhao et al. (2012) stated that the rate of replacement with gaseous CO2 decreased after 10 h of reaction, and the reaction stopped after about 50 h. In the experiments conducted by Ota et al. (2005), they observed that the amount of CH4 hydrate decomposition and CO2 hydrate formation increased rapidly in the early stages (<10 h). This was in agreement with the findings by Hirohama et al. (1996). Subsequently, in the experiments conducted by Wang et al. (2002), the first stage lasted for 5 h; in this stage, the decomposition of the CH4 hydrates and formation of CO2 hydrates was fast. Moreover, Ota et al. (2007) studied cage occupancy during replacement with in-situ laser Raman spectroscopy. They confirmed that both the decreasing rate of CH4 occupancies and the increasing rate of CO2 occupancies were much larger in the earlier (∼10 h described as a rapid surface reaction) than in the later stages. In contrast, Lee et al. (2014) found that the first stage lasted for about 70 h, and the second stage was slow as the resistance to diffusion through the mixed hydrate layer formed increased. Nevertheless, based on the results of the previous studies, it was established that replacing CH4 hydrates using CO2 is feasible, but the replacement rate would decrease after a rapid initial reaction.
In order to improve the replacement percentage and reaction rate, researchers laid emphasis on finding means to mix CO2 into the zone where the CH4 hydrates were present. Qi et al. (2011) studied the mechanism of CH4 guest replacement with CO2 and found that the replacement mainly occurred near the interface. Their conclusion indicated that melting the hydrate once or increasing the interface area would speed up the replacement. Bai et al. (2012) concluded that measures should be taken to eliminate the mass transfer barrier resulting from the formation of the CO2 hydrates layer to increase replacement efficiency. One way is to combine depressurization with CO2 replacement to melt some part of the CH4 hydrates in porous media in order to enhance the CO2 diffusion. The concept of combining depressurization and CO2 replacement was introduced by Zhou et al. (2008a). In their work, depressurization involved decreasing the pressure of sediments (to around 14 MPa) to conditions where CH4 hydrates and CO2 hydrates could coexist. To determine the appropriate conditions for replacing CH4 from hydrates with CO2, it provides a viable solution on in-situ replacement method. However, both CH4 and CO2 hydrates are stable and the barriers between the CO2 molecule and CH4 hydrate layer were not broken under those conditions.
In this study, a new method for enhanced gas hydrate recovery, which combines depressurization and CO2 replacement, is discussed. By incorporating a procedure, in which pressure is decreased to destabilize CH4 hydrates during gaseous CO2 replacement, CH4 hydrates are melted. This new method was experimentally studied to reveal whether it would enhance the replacement reaction. Replacement percentage with and without depressurization were compared.
Experimental section
Apparatus
The adapted schematic of the experimental apparatus is shown in Figure 1. It consists of four parts: reaction cell, a signal conversion device to record pressure and temperature, gas supply system, and mass analyzer to analyze gas composition.
Schematic of the experimental apparatus.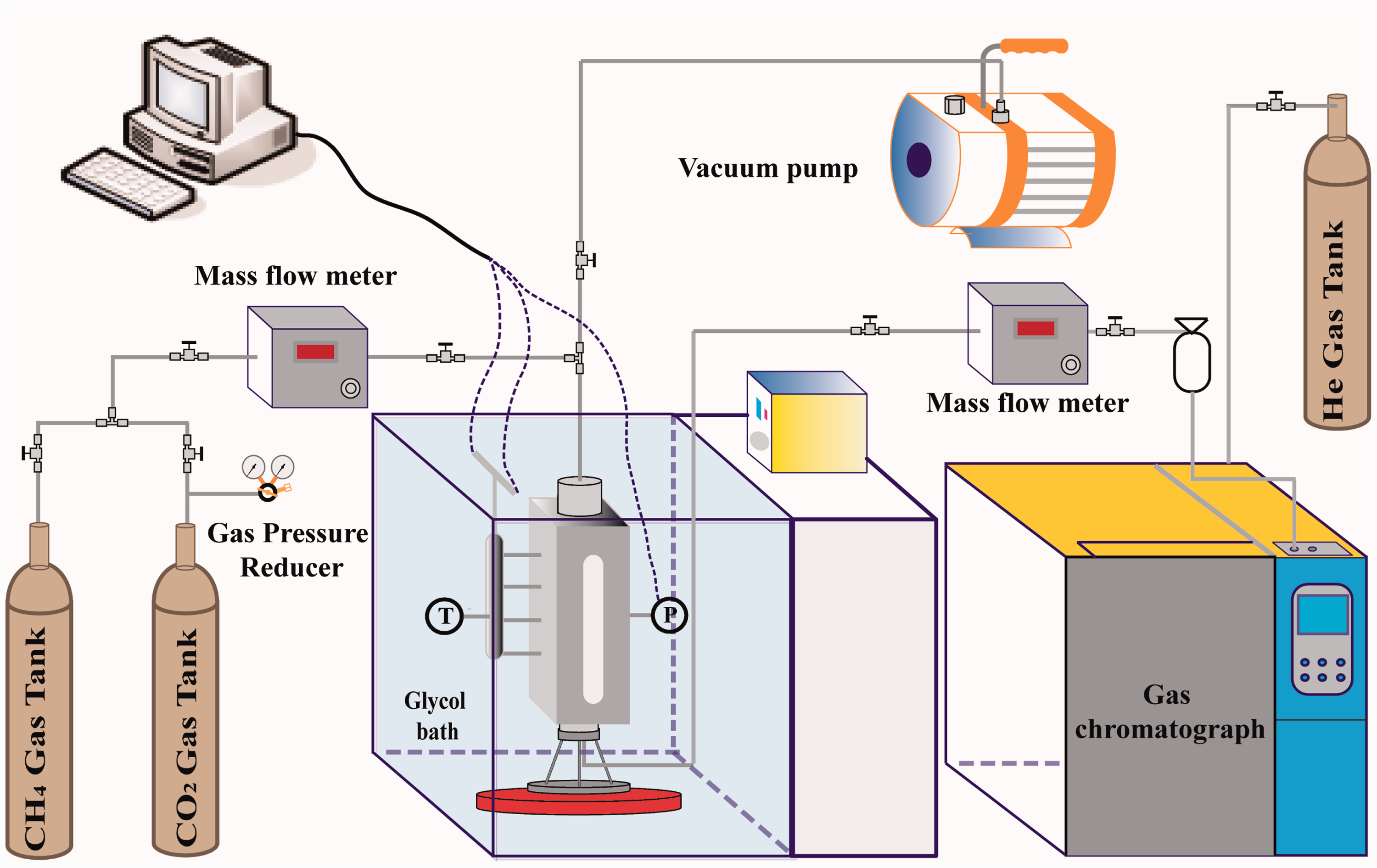
The reactor is made of stainless steel and has a pair of quartz windows on either side for direct observation of the hydrate forming and dissociation processes. The inner volume of the cell is 288 ml with a diameter of 50 mm and height of 130 mm. The temperature of the cell during the experiment was controlled by glycol bath. One pressure and four temperature sensors are attached to the cell. Four temperature thermocouples are distributed at equal distances to record the gradient of the cell temperature. The signals were input into the signal conversion device to record the real-time pressure and temperature conditions.
Procedure
Initially, the reactor was filled with tightly packed quartz sand (BZ08) and uniformly saturated with 20 ml deionized water. Then, a specific amount of CH4 was injected into the cell through a cooler for hydrate formation. The temperature of the bath was set to be 273.65 K. It typically took less than 10 h to form CH4 hydrates under these circumstances. The saturation of the CH4 hydrates was controlled by the amount of CH4 gas injected.
After the formation of CH4 hydrates, the temperature was decreased to 268.15 K in order to reduce the decomposition of hydrates during the process of residual CH4 gas release using a vacuum pump. This temperature was chosen because 268 K is located in the range from 242 to 271 K, where sI hydrates undergo a self-preservation phenomenon releasing the pressure to 0.1 MPa (Stern et al., 2003). Next, chilled CO2 gas was injected to maintain the pressure above the CH4 hydrate equilibrium pressure. The process of gas release and CO2 supply was repeated twice, until the system finally stabilized at the designed pressure such as 3.0 MPa. During the reaction, the pressure varied slightly. The value of the pressure was unified to the pressure recorded after 2 h of reaction, when the diffusion stage was complete. After CO2 gas injection, constant volume replacement reaction proceeded for 15 h with gaseous CO2 at 273 K and 2.8–3.6 MPa. Then, the temperature was set to 268 K in order to reduce hydrate decomposition when the pressure was lowered to 1.6 MPa, which is below the CH4 but above the CO2 hydrate phase equilibrium lines. After the pressure decreased, the temperature was reset to 273 K. Two hours later, cooled CO2 was injected to restore the original pressure (before depressurization).The combined method is designed to increase the diffusion of CO2 when the replacement rate declines.
After 50 h of reaction, the residual mixed gas was released, and the mixed hydrates were decomposed. Samples of the residual and decomposed gases were collected. The sample components were analyzed in the gas chromatogram, the precision of which is 0.01%. From the volume of the collected gas mixture and the percentage of CH4 gas gained by the gas chromatogram, the amount of remaining CH4 hydrates could be calculated.
Models for reaction mechanism
One approach for analyzing the physical mechanisms of the exchange process is to compare the data using physical diffusion/reaction models. The Avrami equation (Avrami, 1939, 1941; Susilo et al., 2007) and shrinking core model (Wang et al., 2002) are well-known physical models for crystallization kinetics. The Avrami model is applicable in the initial stage of the reaction of the hydrate film, while the shrinking core model can be applied to the diffusion through the hydrate film. The general CO2–CH4 exchange has similar physical steps, as it must occur at the surface first and then the diffusion is limited as the interior layers of the hydrate particles are further exposed to CO2. The concepts can be expressed as follows:
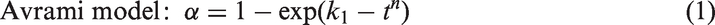
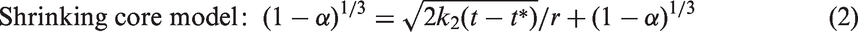
Results and discussion
Combined replacement and depressurization method
Regression parameters of the Avrami and shrinking core models.
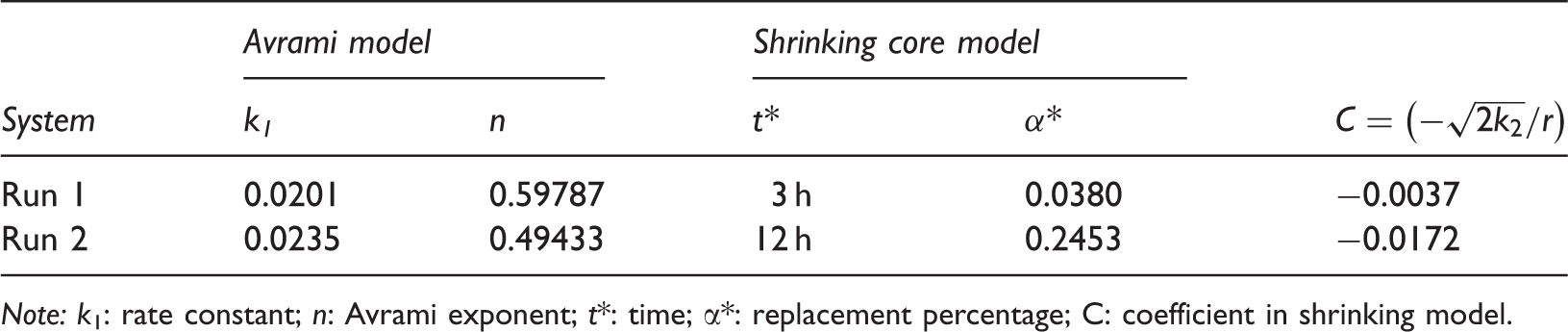
Note: k1: rate constant; n: Avrami exponent; t*: time; α*: replacement percentage; C: coefficient in shrinking model.
Figure 2 shows the replacement percentage trend for the nondepressurization method and fitting line using the two kinetic models (equations (1) and (2)). The Avrami model fits the data from the first 5 h of the reaction; while, the shrinking model best fits the data from 20 to 50 h. During the first 5 h, the amount of CH4 production was more than 60% of the total production over 50 h. The gray zone in Figure 2 is defined as the transition region, where the replacement percentage growth rate drops dramatically.
Replacement percentage comparisons for Avrami (orange line) and shrinking core (blue line) models in Run1. (dots: replacement percentage trend in the first stage, sampling every half an hour; triangles: replacement percent trend in the second stage, sampling every 4 h).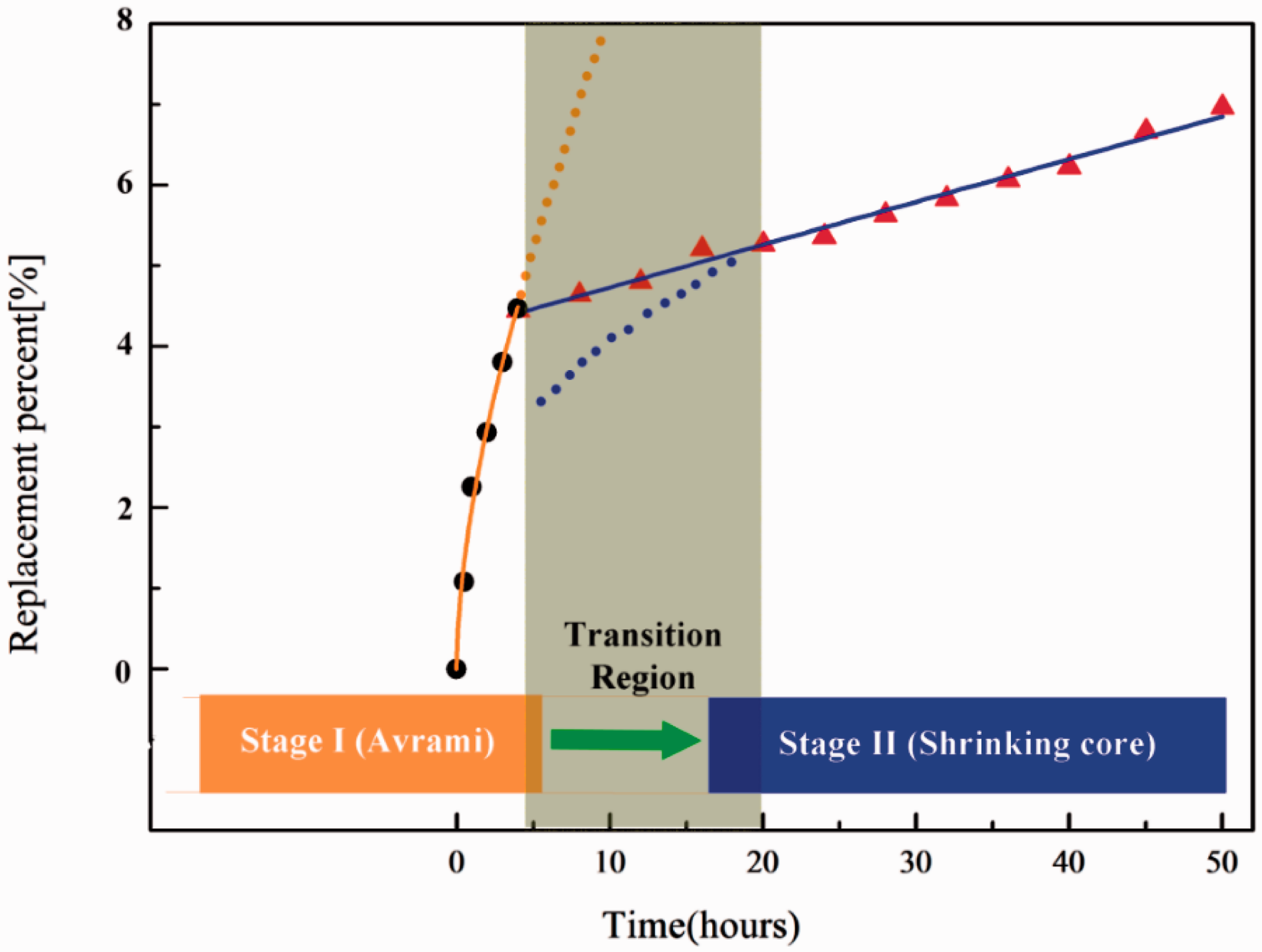
Generally, it was assumed that there were two stages in the exchange process (Lee et al., 2006): (a) fast surface reaction from 0 to 5 h and (b) gradual slow process due to resistance to diffusion through the formation of the mixed hydrate layers from 5 to 50 h. Therefore, the rate-limiting step is the second stage. In this stage, CO2 must penetrate into the inner part of the CH4 hydrate particles; this reaction rate is slow.
In order to improve the replacement rate of the second stage, the combined method including depressurization was proposed. The detailed procedure is described in the Experimental section. The 2-h long depressurization procedure (along the gray zone in Figure 2) was designed to make CH4 hydrates partially melt and provide pathways for CO2 diffusion, as transport of CO2 gas should be fast in porous media (Komai et al., 2002).
Figure 3 shows the replacement percentage data with the depressurization combined method (Run 2) and fitting lines of the two kinetic models. The fitting results were the same as Run 1 during the first 4.0 h (fast surface reaction) and 20–50 h. A complex process followed between 5.0 to 20 h, when results reflected the effects of the 2-h depressurization. During this stage, the process was divided into four parts. (a) During the period from 4.0 to 5.5 h, temperature was decreased to 268.15 K in order to reduce the decomposition of hydrates in the process of depressurization, and pressure was stabilized at 3.1 MPa. The replacement percentage decreased because the lower temperature enhanced CH4 hydrate reformation. (b) At 5.5 h, pressure decreased to 1.6 MPa within a few seconds. The decomposed amount of CH4 hydrates was neglected as pressure and temperature conditions were within the self-conservation range. Then temperature gradually recovered to 273.65 K from 5.5 to 6.5 h. In Figure 3, the replacement percentage was increased rapidly, as the recovery of the temperature would cause the decomposition of CH4 hydrates. This would not be caused by replacement and should be excluded from the total replacement percentage. At the end of temperature recovery procedure, the pressure was 2.1 MPa with T/P condition below methane hydrates phase equilibrium line. (c) During the period from 6.5 to 8.5 h, temperature and pressure were stable at 273.65 K and 2.1 MPa, respectively. The replacement percentage increased. Compared with Run 1 (273.65 k, 3.1 MPa), the higher speed of accumulated replacement percentage in this period indicated that depressurization can enhance the CO2–CH4 exchange process; and (d) during the period from 8.5 to 11 h, pressure increased to 3.1 MPa in a few seconds and temperature was stable at 273.65 K. CH4 hydrates reformed due to the higher pressure. From 11 h onward, the replacement reaction proceeded. The CH4 hydrates that decomposed in the recovery temperature stage, during the period from 5.5 to 6.5 h, reformed. The substantial increase of replacement percentage from 4.64% in 4 h to 25.04% in 11 h shows the degree of the depressurization effect.
Replacement percentage comparisons for Avrami (orange line) and shrinking core (blue line) models in Run 2.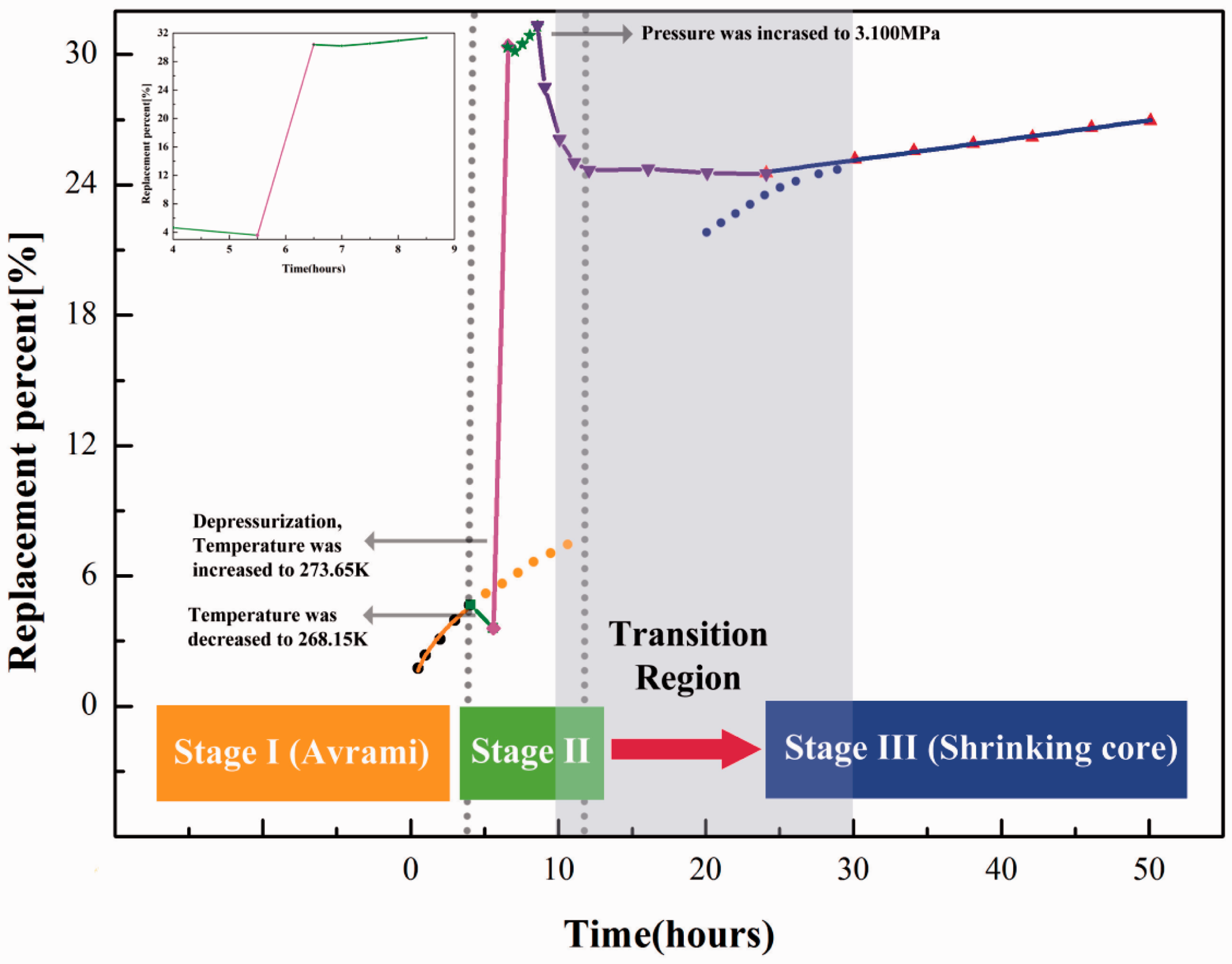
Pressure and temperature trends of CO2 replacement combined with depressurization
Figure 4 shows the typical variation of temperature and pressure for the combining method, starting from the CH4 hydrates formation to the end of the 50-h replacement reaction. The first stage is CH4 hydrate formation from 0 to 200 min, which is indicated by a sharp decline in pressure and an increase in temperature. This is followed by the steady trend in temperature and pressure after CH4 hydrate formation, from 200 to 1400 min. The period from 1400 to 1600 min involves the procedure of temperature reduction to reach the hydrate self-preservation range, before releasing residual CH4 gas. From 1600 to 2400 min, the replacement reaction proceeded for about 13 h at 2.9 MPa and 274 K. At 2400 min, the temperature was set at 268 K to reduce hydrate decomposition when lowering pressure to 1.6 MPa, which is below the CH4 hydrate phase equilibrium line but above the CO2 hydrate phase equilibrium line. After the pressure decreased, the temperature was reset at 274 K. The pressure increased slightly due to the temperature recovery and partially melting CH4 hydrates. Two hours later (at 2700 min), cooled CO2 was injected to obtain a pressure of 2.9 MPa. This stage is located in the stable zone of the CH4 hydrates. After that, the reaction continued in controlled volume until the end of the experiment.
Variation of temperature and pressure in the combined method process.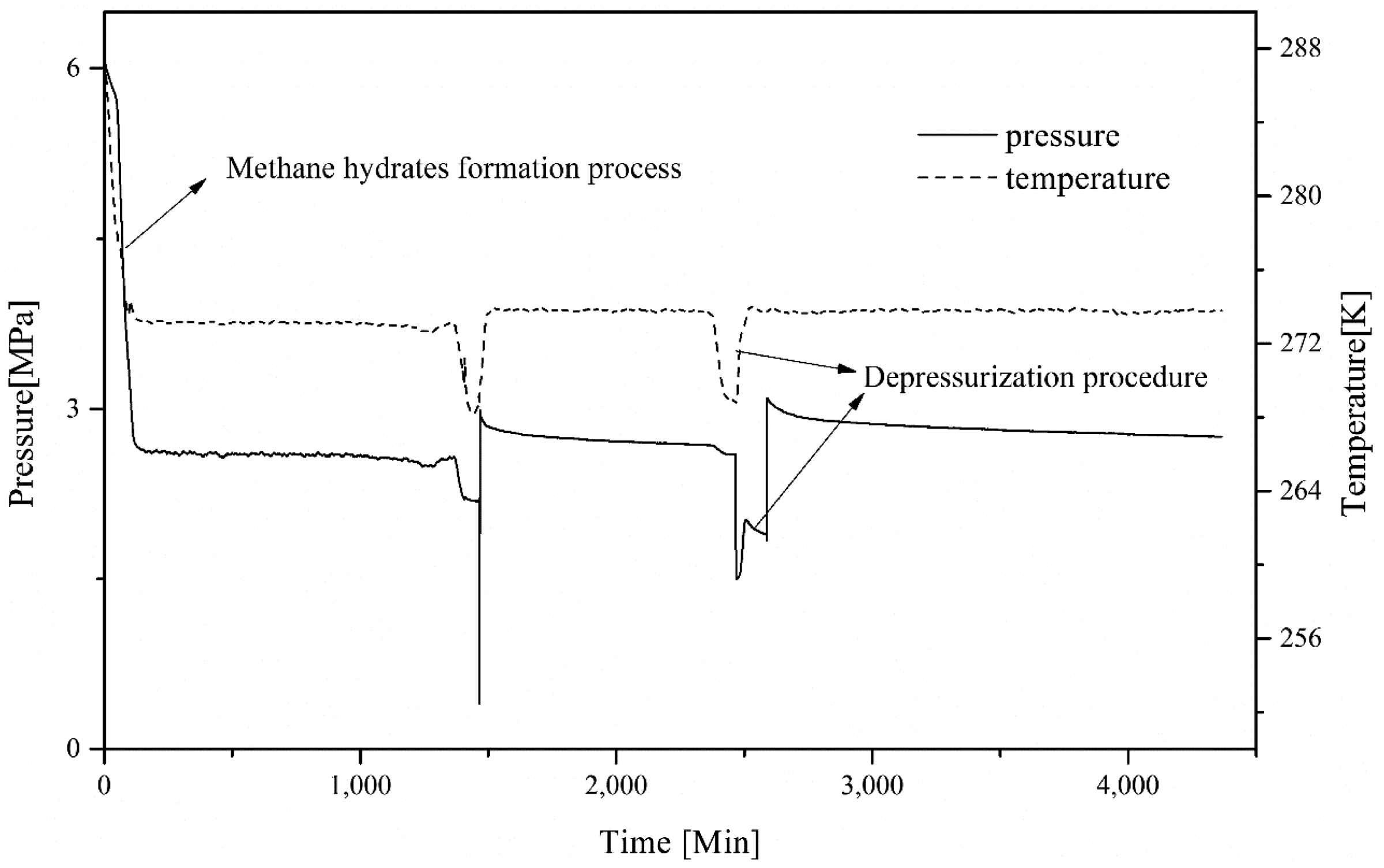
Five groups of experiments for the combined method were conducted, with the same 50-h duration. Temperature and pressure conditions were nearly the same and controlled below the CO2 phase line and above the CH4 hydrates line. CH4 hydrates saturation ranged from 0.11 to 0.22. During the 2-h depressurization procedure, temperature and pressure conditions were located below the CH4 and above the CO2 hydrate lines, which will have caused CH4 hydrates to decompose either by depressurization or heating when the temperature recovered. This part of the CH4 hydrates decomposition was not caused by replacement and should be discussed separately. Thus, the effective replacement percentage is defined as the difference between the percentage of CH4 hydrates decomposing during depressurization and the total replacement percentage.
Replacement percentage with and without depressurization as a function of CH4 hydrate saturation
Experimental data of the depressurization and nondepressurization replacement methods.
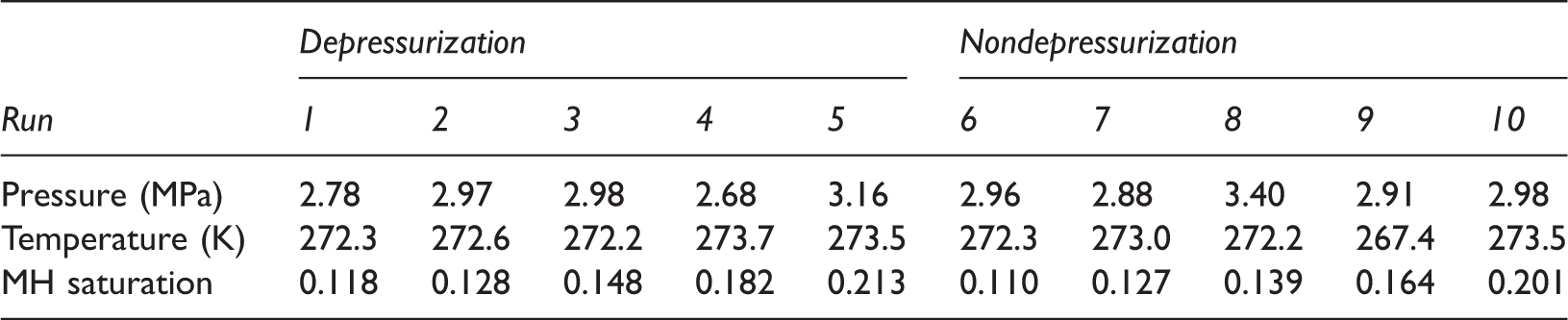
Figure 5 shows the comparison of the replacement percentage of runs with CO2 swap method and combined method. Effective and whole replacement percentage is analyzed.
Comparison of replacement percentage with and without depressurization in 10 runs (dots: replacement percentage during the 50-h replacement reaction; squares: effective replacement percentage gained by excluding decomposed methane hydrates in depressurization procedure over the entire procedure; diamonds: methane hydrates not decomposed by replacement during depressurization; triangles: replacement percentage of runs without depressurization procedure).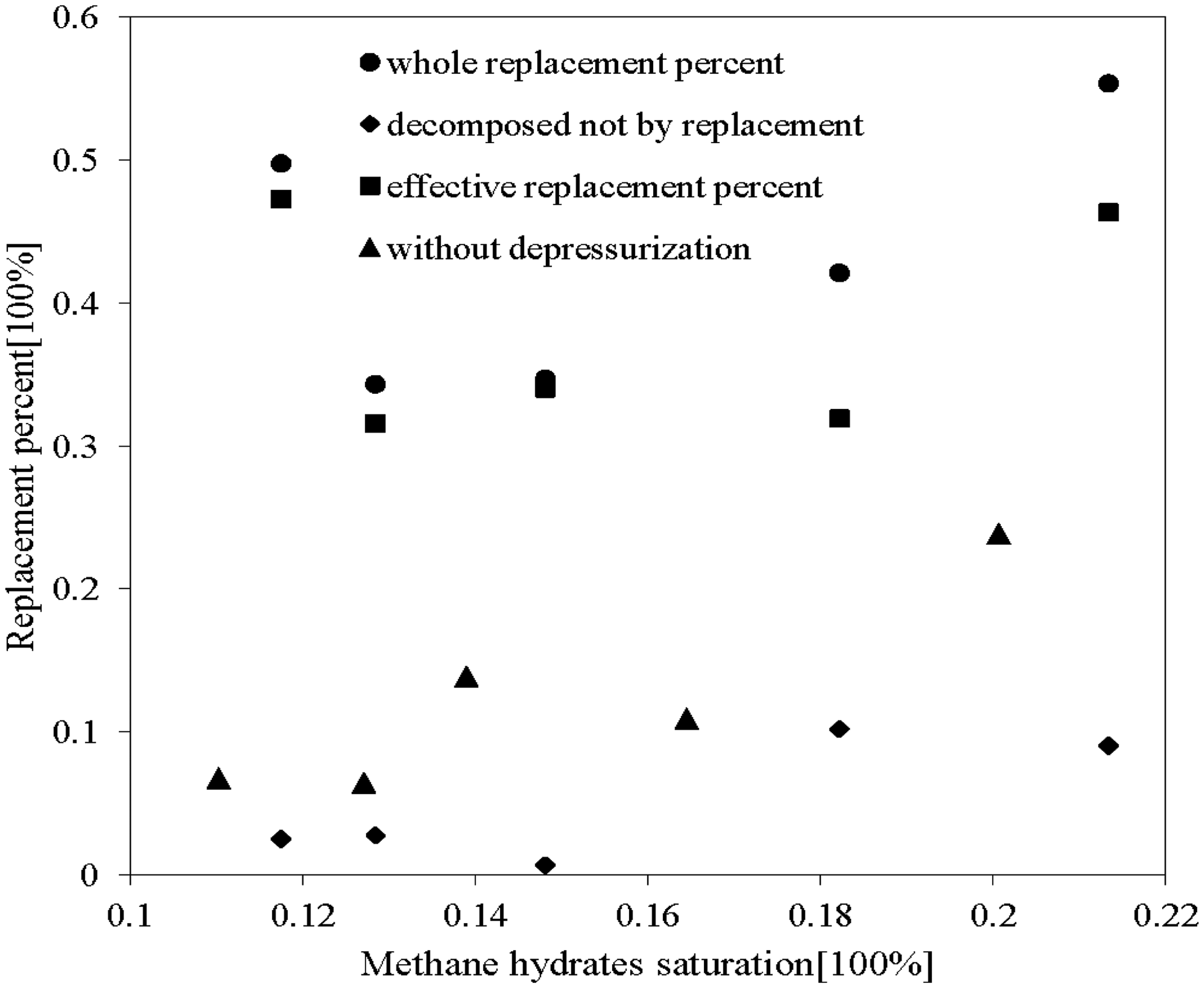
For the combined method (Figure 5), the effective replacement percentage is irrelevant to CH4 hydrate saturation and maintains average levels between 0.30 and 0.48. However, the ratio of the CH4 hydrates decomposed during depressurization is larger for higher CH4 hydrate saturation. It was also found that the effective replacement percentage is much higher than in the trials for CO2 replacement, when the replacement conditions were the same. Replacement percentage of runs using only CO2 replacement were below 0.25, while runs with added depressurization were more than 0.31. The results revealed that the method including depressurization is effective for enhancing CH4 hydrate recovery.
It can be concluded that the 2-h depressurization will result in some CH4 hydrate dissociating and the loosening of porous sediment, which would support the replacement reaction. Larger replacement percentage indicates that higher percentages of CH4 can be replaced, and more CO2 gas can be sequestrated in the form of hydrates. For higher saturated CH4 hydrates, the reaction area between gaseous CO2 and CH4 hydrates is larger, and more CH4 hydrates will decompose in the depressurization procedure, which will lead to larger replacement percentage for both methods.
Conclusions
In this study, a new way to enhance the exploitation of natural gas from CH4 hydrates, combining CO2 replacement and depressurization, was experimentally studied in a high-pressure vessel. An effective replacement percentage was defined using the results of the combined method. Under the same conditions, it was found that the replacement procedure that included depressurization improved the replacement percentage; thus, it was considered effective for enhancing CH4 hydrate recovery. The results supported the hypothesis that the efficient transport of CO2 gas molecules in sediments will effectively promote the replacement procedure. In future research, it would be beneficial to explore the stability of the sediments when using combined methods.