
Abstract
Objective
Autosomal dominant centronuclear myopathy (AD-CNM) is a neuromuscular congenital disease caused by mutations in the DNM2 gene that encodes dynamin-2 (DNM2). The main clinical features of AD-CNM are progressive weakness and atrophy of skeletal muscles. However, cognitive defects have also been reported, suggesting that AD-CNM-causing mutations in DNM2 might also affect central nervous system (CNS). We recently demonstrated that defects in excitatory synaptic transmission occur in the brain of transgenic knock-in (KI) mice harboring the DNM2 p.R465W mutation, the most common causing AD-CNM. As DNM2 regulates the trafficking of glutamate-AMPA receptors (AMPARs), major mediators of excitatory synaptic transmission in mammals, it is feasible that the synaptic availability of AMPAR is affected in the context of AD-CNM. The main objective of this work was to evaluate the impact of the p.R465W DNM2 mutation on the GluA1-AMPAR-subunit synaptic availability in the brain of KI mice.
Methods
We addressed an experimental quantitative study. By using subcellular fractionation and western blot we quantified the expression of GluA1 and synaptic proteins in hippocampal total homogenates and postsynaptic densities (PSDs) in the brain of WT and KI mice. By total internal reflection microscopy (TIRFM) we also analyzed the arrival and residence time of GluA1 into the plasma membrane of hippocampal cultured neurons.
Results
Although we did not observe significant differences in the GluA1 expression in hippocampal total homogenates, it was significantly reduced in the PSDs of KI compared to wild-type (WT) brains. Moreover, the residence time of GluA1 in the surface membranes of KI hippocampal neurons was significantly reduced compared to WT neurons.
Conclusion
These data strongly suggest that the p.R465W mutation in DNM2 perturbs synaptic GluA1-availability in hippocampal neurons, likely leading to defects in excitatory synaptic transmission.
Introduction
Autosomal dominant centronuclear myopathy (AD-CNM) is a congenital myopathy, mainly caused by mutations in the gene encoding dynamin-2 (DNM2).1,2 DNM2 is a large GTP-ase implicated in endocytosis, exocytosis, and cytoskeleton remodelling in different types of cells. 3 Congruently, AD-CNM-causing mutations have been shown to impair these DNM2-dependent processes.2,4,5 Clinically, AD-CNM manifests with progressive weakness and atrophy of skeletal muscles,1,2 causing a wide range of motor defects, including walking, feeding, and breathing difficulties.1,2 Intellectual disabilities have also been observed in AD-CNM patients,6,7 although the molecular mechanisms implicated remain elusive. We recently demonstrated that a knock-in mouse that harbors the most common DNM2 mutation, that is the KI-Dnm2R465W/+ mice (thereafter called KI), exhibits significant impairment in the hippocampal excitatory synaptic transmission and neuronal plasticity. 8 Hippocampal neurons in KI mice show structural defects, including reduced dendritic arborization and diminution in dendritic spine density. 8 As DNM2 has been shown to modulate the insertion and removal of AMPARs to and from synapses, 3 and these receptors are major mediators of the excitatory synaptic transmission in the mammalian brain, 9 the reported defects in excitatory synaptic transmission in KI mice 8 may rely on perturbations in the synaptic availability of AMPARs. Here, we show that the relative amount of GluA1-subunit of AMPAR is not affected in total hippocampal tissue of KI mice compared to WT. However, a significant reduction in GluA1 expression is observed in postsynaptic densities (PSDs) isolated from KI compared to WT hippocampal tissue. Furthermore, the time that GluA1 remains in the neuronal surface membranes of hippocampal neurons is significantly shorter in KI compared to WT neurons. These data strongly suggest that the p.R465W mutation in DNM2 reduces synaptic availability of GluA1 in hippocampal neurons, leading to defects in excitatory synaptic transmission in a murine model of AD-CNM.
Methodology
Animals
Protocols were conducted following standard guidelines for caring experimentation animals, as we previously described. 8 Protocols reported here were carried out with the approval of the Institutional Committee for the Care of Laboratory-Animals (CICUAL) of Universidad de Valparaíso (Number BEA131-18, date of approval August 19, 2021). Considering the “3Rs” principle, we reduced the number of animals by using in this study samples and records coming from the same mice used in our previous report. 8 Therefore, in the current work we did not apply exclusion/inclusion criteria. KI mice and their WT littermates (C57BL/6 strain) were housed at the Institutional Animal Facility of Escuela de Medicina, Universidad de Valparaíso in a temperature- (22 °C) and humidity-controlled Animal Facility under a 12-hour light/dark cycle, with ad libitum access to food and water. Male and female mice were used in all experiments: postnatal (P0–P2 for primary neuron cultures and P5 for Western blot) to 6-month-old KI and WT mice. Genotyping was performed as we previously described 8 by PCR, using DNA extracted from tails and the primers: 3′-CTGCGAGAGGAGACCGAGC-5′ (forward) and 3′-GCTGAGCACTGGAGAGTGTATGG-5′ (reverse). The PCR products were visualized after agarose gel electrophoresis; 445 and 533 bp bands corresponded to WT and DNM2 mutant alleles, respectively.
Subcellular fractionation: PSD-enriched fraction isolation
PSDs-fractions were prepared as we previously described. 10 Briefly, brains were removed, and hippocampi were dissected. Ex vivo tissue was homogenized using a Dounce Grinder in ice-cold (4 °C) homogenization buffer (in mM: 320 sucrose, 4 HEPES, 1 EGTA, and protease and phosphatase inhibitor's cocktail, buffered to pH 7.4). Homogenate was centrifuged at 1000 × g for 10 minutes at 4 °C (Beckman F0630 rotor; IN, USA), and the supernatant was recovered and centrifuged at 33,000 × g for 30 minutes at 4 °C (Beckman S4180 rotor; IN, USA). The pellet was resuspended in the homogenization buffer, layered on the top of a discontinuous sucrose gradient (0.32/1.0/1.2 M), and subjected to ultracentrifugation at 167,000 × g (Himac RPS55T-2 rotor; OE, JP) for 2 hours at 4 °C. Then, samples at the interface of 1.0/1.2 M sucrose were collected, diluted in lysis buffer, and centrifuged again at 33,000 × g for 30 minutes at 4 °C. The pellet was resuspended in a new gradient-loading buffer, loaded on 0.32/1.0/1.2 M discontinuous gradient, and centrifuged at 167,000 × g for 2 hours at 4 °C. The sucrose 1.0/1.2 M interphase containing synaptoneurosomes were delipidated in a delipidating buffer, diluted with a filling buffer to restore the sucrose concentration, and then centrifuged at 33,000 × g for 1 hour at 4 °C. The sediment obtained was washed with 50 mM HEPES–Na and ultracentrifuged at 167,000 × g for 20 minutes at 4 °C. The final sediment corresponded to the PSD-enriched fraction and was resuspended in 50 mM HEPES–Na and homogenized. Total proteins in total hippocampal homogenates and PSD-fractions were quantified using the Qubit® Protein Assay Kit (Invitrogen; Thermo Scientific, IL, USA).
Western blotting
The relative amount of AMPAR-GluA1-subunit was evaluated in total homogenates and PSD-enriched fractions isolated from hippocampal tissue of KI and WT mice. The relative amounts of synaptophysin (SYP) as a presynaptic marker, PSD95 as a postsynaptic marker, and α-tubulin as a loading control were also assessed. Equal amounts of protein (40 µg) were diluted in loading buffer (5×) with bromophenol blue and subjected to 10% SDS-PAGE. Electrophoreted proteins were transferred to polyvinylidene fluoride (PVDF) membranes (BioRad, CA, USA), blocked with blocking solution (EveryBlot Blocking Buffer, BioRad, CA, USA), and immunolabeled with specific antibodies against GluA1-AMPAR-subunit (mouse anti-GluA1; #MA5-27694 Invitrogen; 1:1000), synaptophysin (mouse anti-SYP; #sc-17750; Santa Cruz Biotechnology; 1:1000), PSD95 (rabbit anti-PSD95; #51-6900 Invitrogen; 1:1000) and α-tubulin (sheep anti-α-tubulin; #ATN02 Cytoskeleton Inc; 1:3000). Blots were developed by chemiluminescence using an ECL reagent (Pierce; Thermo Scientific, IL, USA) and detected using the iBrigth-FL1500 Imaging system (Invitrogen, Thermo Fisher Scientific, MLS, SG). The densitometry of immunoreactive bands was quantified using the Image J software (1.54k, NIH, MD, USA).
Hippocampal neuron primary culture
Hippocampal neurons were cultured from postnatal (P0–P2) as we previously described.
8
Briefly, hippocampi from individual pups were dissected, trypsinized for neuron dissociation, and plated in poly-
Total internal reflection microscopy (TIRFM)
To monitor GluA1 arrival to the surface membranes of hippocampal neurons, we used TIRFM. Neurons efficiently transfected with GluA1-SEP were mounted in a recording chamber perfused in Tyrode extracellular solution (mM: 137 NaCl, 2.7 KCl, 1 MgCl2, 1.8 CaCl2, 0.2 Na2HPO4, 12 NaHCO3, 5.5 glucose, pH 7.2) at 37 °C. Live cells were monitored using an inverted microscopy (Nikon Eclipse Ti-E) equipped with a 100×/1.49 NA Plan APO TIRF objective (Nikon) and a Perfect Focus Unit (Nikon, TYO, JP). A surface reflective interference contrast (SRIC; NIKON) was used to identify the areas of neuron membrane that were contacting the glass. SEP was excited by a 488 nm laser (488-20LS, OBIS, Coherent, CA, USA). Spontaneous fluorescent events were recorded at 3.3 Hz for 1 minute with a digital camera (C11440, ORCA-FLASH 2.0; Hamamatsu Photonics, JP) controlled by the NIS-Element viewer 4.3 software (Nikon, TYO, JP). 1-Minute time-lapses were analyzed with the ImageJ software (1.49v, NIH, MD, USA) blind to the neuron genotype. Single fluorescent events were visualized as bright spots in the TIRFM evanescent field. Spots were detected considering spot diameters ranging from 2 to 50 pixels and a change in fluorescence (ΔF) of at least 3 standard deviations above baseline (F0). The number and duration of events were analyzed per each experimental condition.
Statistical analyses
The statistical analyses were performed using the GraphPad Prism 10 software (GraphPad Software Inc., CA, USA). The normality distribution of the data was tested using the Shapiro–Wilk test. The significance of differences was tested by the Mann–Whitney probe for two sample comparisons. Unpaired ANOVA with Tukey’s test was used for multiple comparisons. All data are represented as the mean ± SEM, with a p-value <0.05 being significant. The number of animals (N), statistical test used, and p-values are included in the respective figure legends.
The ARRIVE 2.0 reporting guidelines 11 was considered to conduct and describe our study.
Results
In order to evaluate whether the p.R465W mutation in DNM2 affects AMPAR-expression in brain, we quantified the relative amounts of GluA1-AMPAR subunit in hippocampal homogenates, isolated from postnatal (P5) and adult (6-month-old) KI and WT mice. The synaptic vesicle (SV) transmembrane protein SYP was used as a presynaptic marker, and α-tubulin was used as a loading control. As observed in Figure 1, total expression of GluA1 or SYP was not significantly modified in KI compared to WT brains, neither in neonatal nor in adult mice (Figure 1A–D). To evaluate whether the synaptic availability of AMPAR was affected, we separated PSDs by subcellular fractionation and quantified the relative amount of GluA1 normalized to the PSD-marker PSD95. As shown in Figure 1E–G, GluA1 was significantly reduced in KI compared to WT PSD-enriched fractions, suggesting that the synaptic availability of AMPAR is impaired in KI compared to WT hippocampi. While PSD95 levels were not modified with age in WT PSD-fractions, adult KI mice exhibited a significant reduction in the expression of PSD95 compared to postnatal KI tissues (Figure 1E–G).
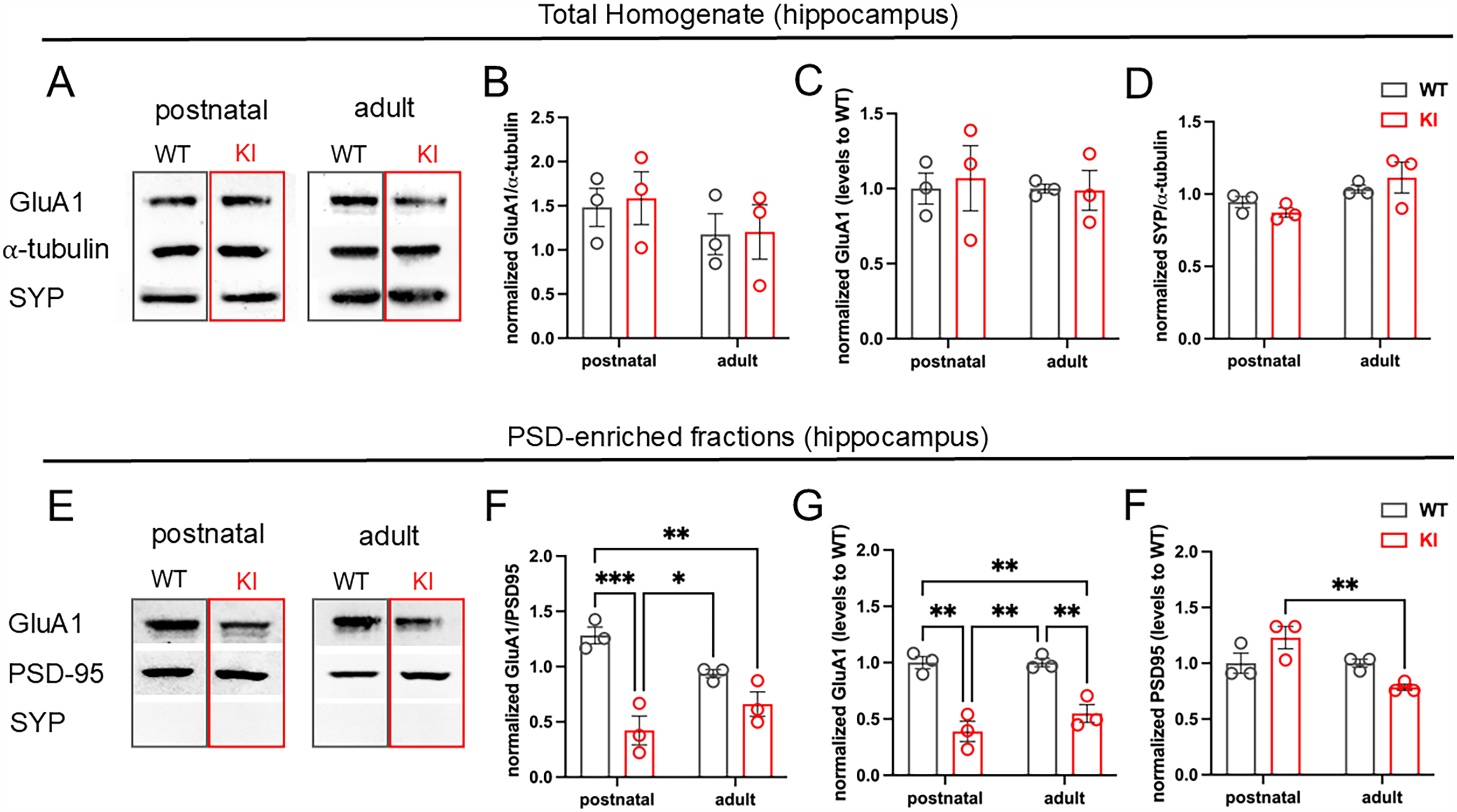
GluA1 expression at the postsynaptic densities is significantly reduced in KI compared to WT hippocampal tissue. Hippocampal homogenates and PSD-enriched fractions from postnatal and adult brains of KI and WT mice were separated by SDS-PAGE, transferred to PVDF membranes, and incubated with specific antibodies against GluA1, SYP, and PSD95 as pre and postsynaptic-markers and α-tubulin as a loading control. (A and E) Representative blots of total hippocampus homogenates (A) and PSD-enriched fractions (E). (B) GluA1 relative amount normalized to α-tubulin in total hippocampal homogenates; (C) GuA1 relative to WT levels; (D) relative amounts of SYP normalized to α-tubulin. (F) GluA1 relative amount normalized to PSD95 in PSD-enriched fractions; (G) GuA1 relative to WT levels; (H) PSD95 relative to WT levels. Grey bars correspond to WT, and red bars correspond to KI. Note that GluA1 is significantly lower in total homogenates and PSDs of KI compared to WT hippocampus. Data represent the mean ± SEM. N = 3 mice per experimental condition. Unpaired ANOVA with Tukey’s multiple comparison tests ***p < 0.001; **p = 0.008; *p = 0.02.
To further evaluate whether synaptic AMPAR availability is affected in the AD-CNM context, we monitored the arrival of GluA1 to the plasma membrane of cultured hippocampal neurons transfected with the construct GluA1-SEP, using TIRFM. With this strategy, the insertion of GluA1 into the surface membranes was monitored as bright fluorescent spots, providing information on the number of receptors arriving at the surface membrane, as well as the time that they remain there. Figure 2A shows representative WT and KI neurons transfected with GluA1-SEP. Figure 2B shows representative kymographs and fluorescence profiles of the bright spots in WT and KI neurons. Although the number of fluorescence spots/per area or the frequency of events/per second were not significantly modified between WT and KI neurons (Figure 2C, D), the duration of GluA1 spots was significantly reduced in KI compared to WT neurons (Figure 2E, F), suggesting a lower stability of AMPAR in the surface membrane of hippocampal neurons in the context of AD-CNM.

The residence time of the GluA1-AMPAR-subunit at the surface membrane is reduced in KI compared to WT hippocampal neurons. GluA1-SEP transfected hippocampal neurons cultured from WT and KI postnatal mice (P0–P2) were visualized by TIRFM to monitor spontaneous fluorescent events. (A) White arrowheads highlight GluA1-SEP insertion events visualized as bright spots. Magnification, 100×. Note that images show the area of the cell that is attached to the glass, where GluA1 spots were monitored and analyzed; Scale bar = 10 μm. (B) Representative ΔF/F0 fluorescence profiles with the respective kymograph of WT (black) and KI (red) GluA1-SEP events. (C) Graph summarizes the mean number of fluorescent events per 1 μm2 of area. (D) Graph summarizes the mean insertion frequency, calculated as the number of events per second. (E) The mean duration per individual event and (F) the mean duration of events normalized per cell were analyzed. Note that the duration of events was significantly lower in KI compared to WT neurons. Data represent the mean ± SEM. N = 6–8 neurons per genotype from 3 to 4 cultures. Unpaired Mann–Whitney test, ***p < 0.001 versus WT condition; *p = 0.02 versus WT condition.
Together, these data strongly suggest that the p.R465W mutation in DNM2 perturbs synaptic AMPAR availability in hippocampal neurons, leading to defects in excitatory synaptic transmission in a murine model of AD-CNM.
Discussion
Dynamins are key modulators of synaptic transmission by controlling SV recycling at the presynapses, as well as the insertion and removal of receptors at the postsynapses. 3 Particularly DNM2, the only dynamin isoform ubiquitously expressed in all the mammalian tissues, 12 plays a presynaptic and postsynaptic role at the central nervous system (CNS). 3 DNM2 participates, in an activity-dependent way, in the SV-endocytic recycling contributing to the maintenance of the readily-releasable pool of SVs. 13 At the postsynapses, DNM2 mediates the insertion and recycling of AMPAR to and from PSDs. 3 This suggests that DNM2 is a key player regulating efficiency of excitatory synaptic transmission in the mammalian brain. We recently reported that hippocampal excitatory synaptic transmission and plasticity are significantly impaired in the brain of knock-in (KI) mice expressing the p.R465W mutation in DNM2, the most common causing AD-CNM. 8 This is a rare congenital disease, mainly affecting skeletal muscles;1,2 however, learning disabilities and cognitive defects have also been reported in AD-CNM patients, suggesting concomitant defects at the CNS.6,7 Our previous report showed that hippocampal slices of KI mice exhibit perturbations in excitatory synaptic plasticity, manifested as a reduction in long-term potentiation (LTP) and long-term depression (LTD), as well as defects in learning and memory. 8 Both, pre and postsynaptic mechanisms, can explain modifications in synaptic plasticity and cognitive functions; 14 hence, the AD-CNM-causing mutation in DNM2 could disrupt its functions at both, pre and postsynapses. Regarding presyanapses, our previous findings showed no changes in the paired-pulse facilitation (PPF) ratio measured through electrophysiological field excitatory postsynaptic potentials (fEPSP). 8 PPF provides an estimation of the neurotransmitter release probability, 8 suggesting that there are no differences at the presynaptic level between WT and KI excitatory hippocampal circuits. Moreover, we demonstrated here that total expression of SYP, a SV-marker, is unaffected in the hippocampus of KI compared to WT mice (Figure 1), strongly suggesting that the AD-CNM-causing p.R465W mutation in DNM2 does not cause presynaptic defects, but rather could affect at the postsynaptic level. With this in mind, we evaluated whether postsynaptic defects could rely on perturbations on AMPAR availability at the excitatory synapses. Our results showed that GluA1-AMPAR subunit is significantly reduced at the PSDs of KI compared to WT hippocampus (Figure 1E–G). The mechanism underlying this effect is not clear, however it is noteworthy that the p.R465W mutation is located in the middle domain of DNM2, critical for its oligomerization and catalytic activity. 8 In this sense, GTP-ase activity of DNM2 has shown to be critical for AMPAR insertion into synapses, by promoting GluA1/2 lateral diffusion from dendritic shafts to PSDs. 15 DNM2 has been also involved in the endocytic recycling of AMPAR from PSDs. 16 Therefore, it is possible that in the context of AD-CNM, the synaptic function of DNM2 is perturbed, impairing AMPAR-trafficking and synaptic availability, leading to excitatory synaptic transmission defects. 8 In this sense, DNM2 can potentially participate at different stages of AMPAR trafficking in neurons, 9 thus, future experiments should be addressed to analyze how AD-CNM-causing mutations in DNM2 could affect not only the availability of the receptor at the neuron surface membranes, but also their impact on AMPAR internalization, endosomal recycling, as well as its localization in synaptic or perisynaptic-sites. In this regard, although the experiments with GluA1-SEP did not show a significant difference in the number nor frequency of events, data show a significant decrease in the time that receptors remained at the surface membrane (Figure 2E, F). In this regard, other DNM2 mutations that cause AD-CNM have been shown to affect duration of exocytotic events in muscle cells 4 as well as slowdown endocytosis in endocrine cells. 5 Indeed, it is feasible that compensatory mechanisms operate in response to DNM2 dysfunctions. For instance dynamin-3, an isoform exclusively expressed at postsynapses,3,11 could enhance its activity in the context of AD-CNM, promoting the endocytic removal of AMPAR from PSDs. Additionally, other subunits relevant for AMPAR-excitatory synaptic activity such as GluA2, have shown to traffic in a DNM2-dependent way, 15 and could be affected by AD-CNM-causing mutations. Future studies should consider to evaluate the impact of AD-CNM-causing mutations in GluA2 synaptic availability and AMPAR function.
Whatever the mechanism, a lower availability of AMPAR at excitatory synapses could lead to perturbations in excitatory synaptic transmission and plasticity, contributing to several neurological disorders. 17 Therefore, a reduction in the expression of AMPAR at the excitatory hippocampal synapses as we observed here, could underly the cognitive defects reported in AD-CNM patients6,7 and in KI-mice. 8
Conclusion
Here we investigated the impact of a DNM2 mutation, linked to AD-CNM, on hippocampal synaptic availability of AMPAR, a major receptor mediating excitatory synaptic transmission in mammalian brains. Our results show for the first time that, in the context of AD-CNM, occurs a significant reduction in the synaptic availability of AMPAR, likely due to defects in DNM2-mediated trafficking to and from synapses. These findings highlight the critical role of DNM2 in maintaining a proper synaptic function at the CNS and strongly suggest that perturbations in AMPAR availability could lead to the neurological defects that has been reported in AD-CNM.
Footnotes
ORCID iDs
Ethical considerations
Authors declare that all the experimental approaches performed in this study were conducted in accordance with international bioethics standards. The protocols reported here were carried out with the approval of the Institutional Committee for the Care of Laboratory-Animals (CICUAL) of Universidad de Valparaíso, Valparaiso, Chile (Number BEA131-18, date of approval August 19, 2021).
Author Contributions/CRediT
CF-M performed experiments and help to write the manuscript; ML-A, MM-A, BG-S, JS-G, LP-V, JA-D, and MGJ-F performed experiments and critically read the manuscript; JAB, MB, and AMC critically revised and edited the manuscript; AOA analyzed and interpreted data and wrote the manuscript; AMG-J conceived the study, designed experiments, analyzed and interpreted data and wrote the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fondo de Equipamiento Científico Tecnológico, Fondo Nacional de Desarrollo Científico y Tecnológico (grant numbers EQM220100, 11180731, 1231511).
Conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
Data supporting the findings of this study are available from corresponding author, upon reasonable request.