
Abstract
Vangl2, a core component of the PCP signaling pathway, serves as a scaffold protein on the cell membrane, playing a crucial role in organizing protein complexes. Cilia, dynamic structures on the cell surface, carry out a wide range of functions. Research has highlighted a bidirectional regulatory interaction between Vangl2 and cilia, underscoring their interconnected roles in cellular processes. This relationship is demonstrated by the localization of Vangl2 at the base and proximal regions of cilia, where it plays essential roles in ciliary positioning, asymmetric distribution, and ciliogenesis. In contrast, the absence of cilia can disrupt Vangl2-mediated signal transduction processes. This review offers a narrative review of recent research on Vangl2's function in cilia and examines the regulatory effects of cilia on Vangl2-mediated signaling.
Introduction
This review reviews the current understanding of the functional role of Vangl2 in cilia and its regulatory influence on Vangl2-mediated signaling. The review identifies common themes in existing research, highlights current trends, and offers recommendations for future studies. Guided by the Scale for the Assessment of Narrative Review Articles (SANRA), this review does not impose specific limitations on time, geography, or research scope. We conducted a comprehensive literature search using databases such as PubMed, Web of Science, and Google Scholar, employing search terms like Vangl2, WNT/PCP signaling pathway, primary cilia, and ciliogenesis. The review synthesizes and analyzes existing studies, ensuring adherence to quality standards in organization, transparency, and analytical rigor. This approach enhances the credibility of the literature review and provides clear guidance for future research directions.
Vangl2 is a scaffold protein of the planar cell polarity signaling pathway
The Vangl2 gene family, first identified over two decades ago in Drosophila as Van Gogh/Strabismus, 1 consists of two homologous genes: Vangl1 and Vangl2. Vangl1 is predominantly located on human chromosome 1q21-q23, whereas Vangl2 is primarily situated on 1p13. 1 Vangl1 and Vangl2 share a 73.1% amino acid sequence homology. 1 These proteins display strong linkage and extensive interactions. As two of the six core proteins in the planar cell polarity (PCP) signaling pathway, they play critical roles in the onset and progression of various diseases. The Vangl2 gene encodes a membrane protein characterized by four transmembrane domains. The N-terminal and C-terminal regions of this protein are exposed to the cytosol. The N-terminus contains two serine/threonine phosphorylation clusters, while the C-terminus features a coiled-coil domain and a PDZ-binding domain, facilitating interactions with several proteins, such as Diego (Dgo) and Disheveled (DVL) (Figure 1). Among these components, Dgo participates exclusively in the PCP pathway, whereas Dvl2 acts as a branching point, playing a critical role in both canonical and non-canonical pathways. Binding with different downstream proteins conferred different biological functions to Vangl2.2,3

Structural organization of Vangl2.
Newly synthesized Vangl2 protein undergoes folding, assembly, and post-translational modifications before being transported from the endoplasmic reticulum to the Golgi complex via coat protein II (COP II). 4 It is then delivered to the cell membrane as part of trans-Golgi network (TGN) vesicles. This process is mediated by interactions with GTP-binding proteins and the Golgi matrix protein attachment complex 1, 5 thereby ensuring an accurate cellular localization of Vangl2.
The PCP signaling pathway is essential for regulating directional cell alignment within tissue planes, affecting cell polarity orientation, guided movement, and epithelial tissue cohesion. This pathway is active in numerous biological processes, including embryonic and nervous system development, hair patterning, and cell migration.
6
As a core component of the PCP pathway, the Vangl2 protein acts as a scaffold, orchestrating physiological processes such as cell migration, adhesion, polarity, and cytoskeleton organization. Through mediating protein-protein interactions and assembling protein complexes, Vangl2 translates spatiotemporal signaling cues into coordinated cellular responses
Cilia are organelles in dynamic equilibrium between assembly and disassembly
Cilia are highly conserved subcellular structures that extend from the cell surface and consist mainly of microtubules. They are composed of several key parts: the basal body, transition zone, axoneme, ciliary membrane, and ciliary tip.18,19 Cilia vary significantly in length, ranging from a few micrometers to 2 millimeters, with a typical diameter of approximately 0.25 micrometers. Excessively long cilia would hinder fluid movement. In mammals, cilia are broadly categorized into motile cilia and primary cilia based on differences in structure and function (Figure 2). Motile cilia are primarily located on the surfaces of the respiratory epithelium, ventricular walls, and epithelial cells of the fallopian tubes. They exhibit coordinated beating patterns that generate fluid movement, essential for cell motility or establishing directional flow across their surfaces. 20 In contrast, primary cilia—often referred to as non-motile cilia—lack motility and function as cellular antennae. These cilia detect physical and biochemical extracellular signals, which they amplify and transduce to coordinate cellular behaviors across temporal and spatial dimensions.21–23 The critical role of primary cilia as coordinators of signaling pathways in development and tissue homeostasis has been well-recognized for over two decades. 24 Primary cilia integrate various signaling pathways, including Hedgehog, Wnt, and PDGFRα, enabling the sensing and transmission of light, mechanical, and chemical signals. In response to mechanical loading, cells such as collecting duct cells, embryonic kidney epithelial cells, and chondrocytes adjust ciliary length through feedback control mechanisms in signaling pathways localized to primary cilia, such as polycystin1/2, cAMP/Ca2+, and Notch.25,26 As environmental sensors, primary cilia play a crucial role in coordinating cellular signaling pathways, developmental processes, and tissue homeostasis, with significant implications for various ciliopathies.
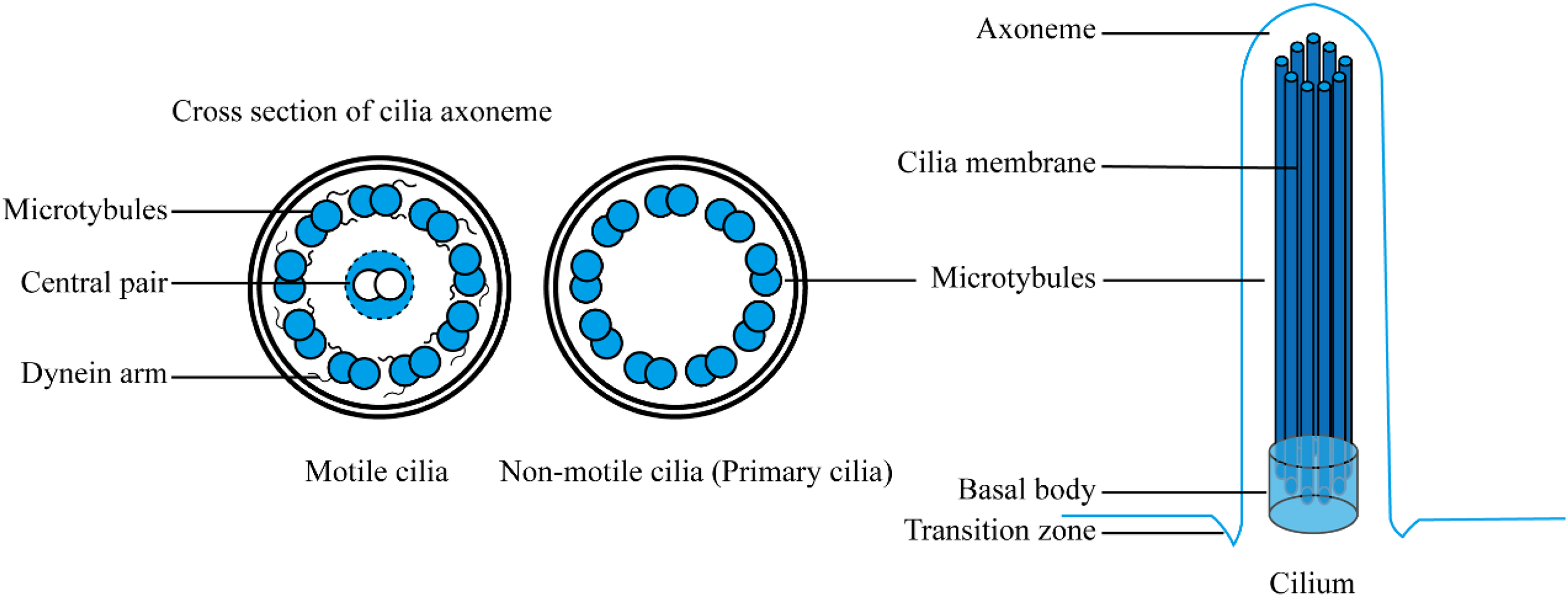
The architecture of cilia.
Ciliogenesis is the orderly process of cilia assembly. Initially, the basal body, derived from the mother centriole, migrates to the cell surface and anchors to the actin-rich plasma membrane. During this stage, the basal body interacts with membrane vesicles, which fuse with the plasma membrane to form the ciliary membrane. The positioning and orientation of the basal body are crucial, as they dictate the direction and sequence of cilia formation. Axonemal microtubules then assemble from the basal body, which functions as the microtubule-organizing center, extending beneath the elongating membrane to facilitate cilia formation. Axonemal microtubules begin assembling from the basal body, which serves as the microtubule-organizing center, extending beneath the growing membrane to support cilia formation. During axoneme elongation, various microtubule-associated proteins are transported into the cilium from the cytoplasm via a movement-dependent, bidirectional transport system known as intraflagellar transport (IFT). 27 Cilia are organelles that maintain a dynamic balance between assembly and disassembly. The process of ciliary disassembly necessitates the destabilization and depolymerization of axonemal microtubules, which involves the Kinesin-13 family proteins, specifically Kif2a and Kif24.28–30 These proteins are activated by proliferative signals, driving both microtubule depolymerization and ciliary disassembly. The activation of Kif24 is regulated through cell cycle-specific phosphorylation, linking it to ciliary disassembly prior to mitosis. Meanwhile, Kif2a is localized at the proximal ends of both centrioles and the sub-distal appendages of the mother centriole and becomes activated via phosphorylation by the G2/M phase kinase PLK163. Histone deacetylase 6 (HDAC6) deacetylates cortactin, which enhances actin polymerization and promotes ciliary disassembly. Polo-like kinase 1 (PLK1), a G2/M phase kinase recruited to the pericentriolar matrix before mitosis, interacts with and activates HDAC6 to support ciliary deacetylation and resorption.31,32 Additionally, casein kinase 1 (CK1) contributes to primary cilium disassembly by phosphorylating DVL2 at S143 and T224. Phosphorylation at S143 of DVL2 is crucial for cilium disassembly, whereas phosphorylation at T224 has a comparatively minor role. 33
Microtubule proteins at the distal ends of cilia undergo constant assembly and disassembly, maintaining a dynamic equilibrium. As a result, both ciliary assembly and disassembly are active processes, and disruptions in this balance can lead to abnormally long or short cilia, or even complete ciliary loss, ultimately impairing cellular function. 34
Vangl2 is essential for the establishment of ciliary polarity and ciliogenesis
Vangl2 is also essential for the retrograde and asymmetric positioning of cilia. 35 It also regulates the positioning of the ciliary basal body and supports microtubule assembly, ultimately influencing axoneme formation.25,36
The influence of Vangl2 on ciliary polarity
Jussila et al. demonstrated the establishment and maintenance of Vangl2 polarization at the basal body through protein degradation methods, highlighting its role in the intercellular propagation of PCP polarity. 37 Additionally, research has shown that Cofilin 1, a muscle actin-severing protein, works synergistically with Vangl2 to regulate PCP in early mouse embryos. 38
Planar polarity is crucial for the posterior positioning of cilia on mouse node cells, which is necessary for establishing left-right asymmetry. In double mutants of Vangl2 and Cofilin 1, cilia remain centralized, resulting in randomized left-right asymmetry. Additionally, Vangl2 mutants in the nasopharyngeal region of mice show defective convergence extension, significant misalignment of ciliary cells along the longitudinal axis, and a loss of tissue polarity. 39 Similarly, Vangl2 has been identified to control the posterior tilt of primary motile cilia within the neural tube, Kupffer's vesicle, and pronephric duct epithelia in zebrafish. It is crucial for the asymmetric positioning of cilia on the apical membrane of neuroepithelial cells.37,40 In mammals, the hearing and sensory organs, specifically the Corti organs, consist of sensory hair cells with uniformly oriented stereocilia that display PCP aligned with the sensory epithelial cells.41–43 Mutations in Vangl2 lead to a shortened, widened cochlea and misaligned stereocilia. The PCP pathway coordinates the uniform orientation of cellular structures and movements within the tissue plane. 44 Additionally, studies indicate that Vangl2 modulates ciliary positioning by interacting with other proteins within the PCP pathway. Further studies suggest that Vangl2 regulates ciliary positioning by interacting with other proteins in the PCP pathway, with support from actin filaments (Figure 3). Specifically, Vangl2, Celsr1, and Frizzled work together to position primary cilia in radial glial cells and guide the directional movement of ciliary clusters on the ependymal membrane. When Celsr1, Frizzled3, or Vangl2 is inactivated, the tissue polarity of multiciliated radial glial cells is disrupted, impairing the establishment of both rotational and translational polarity in ependymal cells. Moreover, Celsr2, Celsr3, Frizzled3, and Vangl2 regulate single-cell ciliary polarity, which is essential for organizing the retina's internal structure. The disruption of tissue organization in multiciliated cells occurs when these four proteins are inactivated at the single-cell level.45,46 This indicates that Vangl2, along with its associated proteins, has a dual role in ciliary organization, operating at both the single-cell and tissue levels. It ensures the proper positioning of primary cilia, which helps align the polarity of individual cells with that of neighboring cells.

The influence of Vangl2 on ciliary polarity. The asymmetric distribution of core component complexes mediated by Vangl2 establishes planar cell polarity (PCP). The signaling of the latter affects the distribution, assembly, and attachment sites of actin, thereby influencing the polarized localization of cilia.
Moreover, Vangl2 shows a temporary asymmetric distribution throughout the ciliary orientation process on the ependyma. This unique positioning allows it to connect fluid dynamics to the spatial organization of cilia, directly linking external hydrodynamic stimuli to intracellular PCP signaling pathways. 47 Vangl1 and Vangl2 are broadly expressed during the formation of left-right (L-R) patterning. Their protein levels are specifically elevated in cells of the posterior neural tube, where they display asymmetric localization along the anterior-posterior (A-P) axis. Knocking out Vangl1 and Vangl2 in Drosophila results in random ciliary positioning around posterior midgut cells and creates turbulent nodal flow, which disrupts ciliary left-right (L-R) asymmetry. 48 Research by Borovina et al. highlights the critical role of Vangl2 in establishing directional, ciliary-driven fluid flow. 40 High levels of Vangl2 inhibitors interfere with basal body orientation. In radial glial cells, hydrodynamic forces at the apical surface help guide PCP proteins to specific locations within the ventricular epithelium. Mechanical sensory proteins polycystin-1 (PKD1), PKD2, and primary cilia in radial glial cells are essential for the correct asymmetric localization of PCP protein Vangl2 in the apical region of ventricular cells. Moreover, the presence of PKD1 or PKD2 directly or indirectly affects Vangl2 levels at the apical surface of these cells. 49 Thus, it is plausible to suggest that PCP proteins play a significant and wide-ranging role in the asymmetric positioning and orientation of primary cilia, which is crucial for establishing directional fluid flow essential to both normal embryonic development and certain diseases.
The role of Vangl2 in the regulation of ciliogenesis
In mammals, motile cilia are present in various organs, including the fallopian tubes, respiratory tract, and brain ventricles. The cilia on ependymal cells in the brain ventricles help regulate cerebrospinal fluid flow. Degradation of Vangl2 disrupts cilia formation and can lead to idiopathic scoliosis, with the severity of spinal curvature potentially correlating with the extent of Vangl2 loss. 37
Vangl2 has also been implicated in the regulation of ciliogenesis, with multiple studies indicating its localization to the basal body and potential involvement in actin organization. 7 In zebrafish, BBS8 plays a critical role in maintaining the typical length of cilia in Kupffer's vesicles, though it does not influence cilia quantity. In contrast, homozygous Vangl2 mutants display reductions in both cilia number and length. Remarkably, when Vangl2-mutant embryos are injected with BBS8-MO alone, cilia numbers decrease even further to 30% of normal levels. These findings indicate that Vangl2 knockdown in zebrafish embryos results in shorter and fewer cilia within Kupffer's vesicles. This suggests a collaborative role between BBS8 and Vangl2 in regulating both cilia length and number in this structure. The physical interaction between BBS8 and Vangl2 has been confirmed by co-precipitation experiments, supporting their functional partnership under physiological conditions. 50 Knockdown of Vangl2 in Xenopus laevis significantly reduces ciliary number. However, co-injection of synthetic Vangl2 mRNA that lacks morpholino-targeted sequences effectively restores the phenotype. 51 This indicates that Vangl2 loss has a direct impact on ciliary assembly.
Research has shown that most core PCP proteins work in conjunction with Vangl2 to regulate ciliogenesis.52–54 Vangl2 and Wtip jointly regulate renal ciliogenesis, and loss of either protein results in renal cysts similar to those seen with ciliary loss. 55 Research has shown that DVL and Vangl2 control the planar orientation of basal bodies, thereby directing the polarized beating of motile cilia. 31 Further studies indicate that DVL and Vangl2, together with other PCP proteins, regulate the directional beating of multiple cilia in mouse brain and zebrafish kidney-pronephric cells. 56 Our research has demonstrated that Vangl2 co-localizes with the basal body of primary cilia in human umbilical vein endothelial cells (HUVECs).34,57 Additionally, both the expression and cellular localization of Vangl2 are affected by varying shear stress levels. 34 Manipulation of Vangl2 expression, through either silencing or overexpression, influences ciliogenesis under different fluid shear stress (FSS) conditions.Vangl2 facilitates primary cilia disassembly by upregulating DVL2 and enhancing the interaction between DVL2 and KIF2A. Concurrently, it promotes primary cilia assembly by downregulating pDVL2 and upregulating proteins essential for ciliogenesis, such as BBS8 and IFT88. This dynamic balance ultimately determines the occurrence of primary ciliogenesis (Figure 4). These findings suggest that Vangl2 is critical in modulating the equilibrium between ciliary assembly and disassembly Additional studies have confirmed that Vangl2 regulates ciliogenesis through its interaction with ciliary assembly-related proteins, including BBS and ARL13B.17,50 Considering BBS8's role in IFT assembly, 58 Vangl2 likely supports ciliary assembly during development by regulating both BBS8 and ARL13B. Additionally, the Wnt5a–CK1–DVL2–PLK1 pathway is involved in ciliary disassembly, 31 with Vangl2 interacting with CK1 and DVL2. Therefore, it can be inferred that Vangl2 is closely associated with ciliary disassembly. Previous studies indicate that Kif2a interacts with both DVL2 and Vangl2 in cells overexpressing Vangl2. Based on this, we propose that Vangl2 might inhibit ciliary disassembly through its interaction with Kif2a.

The role of Vangl2 in the regulation of ciliogenesis. Vangl2 promotes the disassembly of primary cilia by upregulating DVL2 and enhancing the interaction between DVL2 and KIF2A. Meanwhile, it increases the assembly of primary cilia by downregulating pDVL2 and upregulating the assembly-related proteins BBS8 and IFT88. The balance between these two processes ultimately determines the occurrence of primary cilia.
The relationship between Vangl2 and ciliopathies
Ciliopathies are human diseases caused by defects in cilia.36,59 These defects in cilia assembly and signaling are linked to all ciliopathies, which impact nearly every organ system. Mutations in PCP genes can lead to various ciliopathies due to the close relationship between PCP and ciliary function. 60 The range of these diseases includes relatively common conditions like polycystic kidney disease (PKD) as well as rarer genetic syndromes. These syndromes have a wide clinical spectrum, affecting multiple organ systems such as the eyes, kidneys, nervous system, skeletal system, liver, heart, and gonads. 17 Phenotypes associated with ciliary dysfunction in mammals are diverse. They include randomization of the left-right body axis, abnormalities in neural tube closure and patterning, skeletal defects such as polydactyly, cystic kidneys, liver and pancreatic diseases, blindness, loss of smell, behavioral and conduct disorders, cognitive deficits, and obesity.61–63 Research has demonstrated that various ciliopathy genes work together to influence the severity of these phenotypes across species, from worms to humans.64–67 To date, no core PCP genes have been identified as pathogenic or modifier alleles for human ciliopathies. This likely reflects the fact that such mutations would be incompatible with life. However, increasing evidence from animal studies suggests that PCP components regulate ciliary function, implying that PCP genes could be potential candidates.
The interactions between ARL13B and Vangl2 establish new links between cilia and the PCP pathway. ARL13B and Vangl2 form a complex, and their interactions worsen ciliary defects in photoreceptors, influencing the length of photoreceptor cilia. Mutations in ARL13B and Vangl2 result in shortened cilia and outer segments of photoreceptors, which can lead to Joubert syndrome, an autosomal recessive genetic ciliopathy. Similarly, defects in ciliary and basal body functions are associated with ciliopathies, causing the formation of renal cysts. 68 Research has identified cell division defects in cystic kidneys in zebrafish. Knockdown of Vangl2 leads to the formation of renal cysts along with malformations of the pronephric duct.55,69 This phenotype closely mirrors that of Wtip mutants, a member of the Ajuba/Zyxin family of LIM domain proteins. Knockdown of Vangl2 significantly exacerbates cyst formation and pronephric duct malformations in Wtip knockdown zebrafish. This finding underscores the critical role of Vangl2 in coordinating normal pronephric duct formation in zebrafish and its importance for renal branch morphogenesis. 70 It highlights the intricate molecular mechanisms underlying ciliopathy-associated phenotypes and identifies a new role for Wtip in ciliopathies. Specifically, Wtip contributes to the development of anterior and middle pronephric cysts and cell division abnormalities, as well as the link between pronephric duct malformations and the PCP pathway. This indicates an interaction between the two basal body proteins, Wtip and Vangl2. The proper orientation of the mitotic spindle, the morphogenesis of the pronephric duct, and the PCP pathway in the anterior and middle pronephric regions of zebrafish all depend on Wtip. This dependency may play a role in the molecular pathogenesis of renal cysts. Mislocalization of Vangl2 disrupts the localization of other core components of the PCP pathway and interferes with the interactions between Vangl2 and its associated proteins. This disruption could be a contributing factor to diseases caused by Vangl2 gene mutations.
The role of primary cilia in Vangl2-mediated PCP pathway signaling
The primary role of primary cilia is in cellular signaling, as they house various receptors, ion channels, transport proteins, and their downstream effectors. 71 Signaling through cilia orchestrates essential processes in development and tissue homeostasis, such as cell migration, differentiation, cell cycle re-entry, regulation of the cleavage plane during cell division, and apoptosis. Primary cilia can respond to sensory inputs, including mechanical stimuli (like ciliary bending) and chemical signals (such as specific ligands, growth factors, hormones, or morphogens). 68 In certain situations, primary cilia can also respond to light, 72 temperature, 69 osmotic pressure, 73 and gravity. 74 Different tissues feature specific sets of receptors localized to the ciliary membrane, enabling primary cilia to detect diverse environmental signals. Vertebrate PCP proteins are crucial for regulating the polarization of both epithelial and non-epithelial cells, influencing processes such as convergence extension, lung branching, and the orientation of cilia, hair follicles, and inner ear hair cells. 75 Mutations in PCP proteins can lead to conditions like hydrocephalus, laterality defects, NTDs, and impairments in the kidneys, heart, and hearing.
The signaling pathways coordinated by primary cilia are highly diverse and dependent on cell type, with various receptors or channels present in the same cilium either simultaneously or at different times. These pathways include signaling through Ca2+, receptor tyrosine kinases (RTKs), Hedgehog (Hh), Wingless (Wnt), neurotransmitter and purinergic receptors, and communication with the extracellular matrix (ECM).76–85 Notably, the Wnt/PCP signaling pathway, which is localized to cilia, is closely associated with many proteins involved in ciliogenesis. 25 Cilia play crucial roles in Wnt/PCP signal transduction, and defects or abnormalities in cilia are linked to the disruption of Wnt/PCP signaling and ciliopathy-related diseases.61,62,71,86 Research on the ciliary protein OFD1, which is localized to the ciliary basal body, has highlighted the connection between cilia and the Wnt/PCP signaling pathway. 87 Additionally, studies on other ciliary proteins, such as Inversin and NPHP-3, 88 along with the identification of IFT88, Kif3a, and BBS mutants, 89 emphasize the essential role of primary cilia in regulating Wnt/PCP signal transduction. This research underscores the strong association between Vangl2, a core protein of the PCP pathway, and cilia. 71
The initial connection between cilia and the Wnt/PCP pathway was identified through the nephrocystin2 protein (also known as Inv). This protein localizes to cilia and physically interacts with DVL, a core component of the Wnt/PCP pathway.90–92 DVL plays a role in both canonical and non-canonical Wnt signaling. Inv shares significant homology with the fruit fly PCP gene Diego, 93 and its loss leads to convergence extension defects, highlighting its role in vertebrate PCP. Co-transfection experiments have shown that Inv inhibits DVL's ability to activate Wnt-responsive reporter constructs 92 (Figure 5). More than half of the NPHP proteins (specifically NPHP1, 4, 5, 6, and 8) localize to the transition zone at the base of cilia. Here, they play crucial roles in ciliogenesis and in the transport of specific proteins into the ciliary compartment.94–99 Based on these findings, Simons et al. 92 propose that Inv/NPHP2, located at the base of primary cilia, may function as molecular switches to regulate the balance between classical Wnt signaling and Wnt/PCP signaling.

The role of primary cilia in Vangl2-mediated PCP pathway signaling. The abnormal state of cilia affects the expression of Vangl2 at the base, which binds to the signaling proteins Prickle and inversin, resulting in reduced activity of DVL and Dsh. This leads to the suppression of the transmission activity of the planar cell polarity (PCP) signaling pathway.
In epithelial tube cell cultures, primary cilia bend in response to fluid shear or mechanical stimulation, resulting in an increase in intracellular Ca2+ levels.100–102 This indicates that ciliary mechanotransduction is crucial for the normal function of renal epithelia. Loss of cilia can disrupt this process, leading to PKD. Additionally, primary cilia in radial glial cells may regulate PCP signaling transduction in ependymal cells by controlling the asymmetric localization of PKD1 and PKD2. These proteins form a complex in primary cilia, acting as mechanosensors that respond to fluid movements through renal tubules by initiating calcium signals. 93 Loss or mutation of these ciliary proteins can lead to cyst formation. Another ciliopathy gene, OFD1, also causes convergence extension defects in zebrafish and exhibits functional synergy with Vangl2. 103
In summary, these studies highlight the roles of cilia and cilia-related proteins in the Vangl2-centered PCP signaling pathway. However, a systematic approach to fully integrate the role of cilia within the existing framework of PCP signaling transduction mechanisms is still lacking.
Conclusion
In summary, we have elucidated the bidirectional relationship between Vangl2 and cilia. Vangl2 is located at the base of the cilium, where it interacts with various proteins involved in ciliary function. It significantly influences cilia by modulating their assembly, disassembly, maintenance, localization, and overall function through structural modifications. Therefore, investigating cilia with Vangl2 as a foundational element, along with other core proteins of the PCP signaling pathway, offers a valuable and innovative research approach.
Moreover, primary cilia have been documented to play a role in the Vangl2-centered PCP signaling pathway. Deficiency or mutations in cilia-associated proteins can disrupt the Wnt/PCP signaling pathway. Thus, primary cilia are crucial for the transduction of the Wnt/PCP signaling pathway. Further exploration of the interplay between cilia and the Wnt/PCP signaling pathway will help clarify the regulatory mechanisms underlying ciliopathy progression at both the cellular and molecular levels.
Footnotes
Acknowledgments
The authors would like to thank the Department of Biochemistry at Zunyi Medical University for their academic insights. This work was supported by the National Natural Science Foundation of China (NSFC-32260156), and the Science and Technology Foundation of Guizhou Province (Qiankehejichu-ZK [2022] zhongdian 050). They also appreciate the insightful feedback provided by the anonymous reviewers, which greatly contributed to improving the clarity and depth of this review.
Author contributions
Huanyong Qin, Ting Liang and Chuanfen Zhang performed the literature search. Huanyong Qin and Junlin Wu wrote the manuscript. Huanyong Qin and Xin Sheng revised the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical considerations
The authors declare that all data and figures included in this review were obtained from publicly available sources, and proper attribution is given to the original authors and copyright holders.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the National Natural Science Foundation of China (32260156), and the Science and Technology Foundation of Guizhou Province (Qiankehejichu-ZK [2022] zhong dian 050).