
Abstract
The Michaelis constants derived for two reversible uni-reactant - uni-product reaction models, given originally by Haldane, are corrected. In the direction starting with the reactant having the lower binding constant, the steady state is one in which the enzyme-reactant intermediate has a concentration approximating the final equilibrium concentration., Consequently, the Haldane relationship is generally invalid, and kinetic analyses to validate the use of the kinetic constant ratio (kcat/Km) as a measure of specificity are also generally invalid. This correction of Michaelis constants is pertinent to attempts to back calculate rate constants from experimental values: the Michaelis constant used must be correct.
Keywords
Introduction
Haldane1,2 applied the now-called (quasi) steady-state assumption introduced earlier by Briggs and Haldane
3
to the reversible reactions shown in Figure 1, and derived for both models, and for the reactions in both directions, Henri-Michaelis-Menten equations. These, for a reaction starting with reactant A, apply to initial steady state velocities, have the form shown in equation 1, and identified the kinetic constants,

Two models for(A)—uni-product (B) reaction catalysed by an enzyme E. The corresponding small case letters represent concentrations,and the letters k are rate constants.
Since roughly the middle of the last Century the view has been held that experimentally-derived kinetic constants should be viewed as purely empirical measurements which under defined conditions characterize the enzyme. Their values are now obtained directly, using computer-aided numerical methods,
9
from primary experimental data (reactant concentrations measured at intervals) rather than from secondarily derived reaction rates. None the less, the derivations of Haldane still appear in current texts and in the literature, and furthermore so do other relationships which are based on his work. Haldane's own relationship1,2 between the kinetic constants of the forward and reverse reactions and the equilibrium constant of the reaction, and kinetic demonstrations that the ratio of kinetic constants, kcat/Km, is a good measure of enzymic specificity10,11 are not generally valid. Recently, it has also been demonstrated
12
that methods of numerical integration can be used to obtain rate constants from a measured Km. Such an exercise requires the correct structure of the Km, written in terms of the rate constants of the particular kinetic model used. In the case cited, an irreversible form of model 1 was used with the correct expression for the Km, but for, any extension of the procedure to reversible reactions, the present work should be viewed as a cautionary note.

Quasi-Equilibrium in the steady state, and the correct Michaelis constants for model 1
Morales and co-authors4,5 pointed out that, for x to pass through a maximum value when dx/dt = zero, d2x/dt
2
must be negative at the maximum (which is the value used as the steady-state concentration). They showed that, when starting with A, this required that k1 > k-2. The implication of this is that, when k-2 > k1, x rises only to xequ. (The analysis of Morales and co-authors is essentially repeated in appendix i, where I use their approach as part of the analysis of model 2). The demonstration that, in the pre-steady state, x rises monotonically to xequ is provided by the work of Miller and Alberty,
6
Walter and Morales
7
and Tzafriri and Edelman.
8
When the pre-steady state is brief (Briggs and Haldane's “in the first instant of the reaction”,
3
) and if the product concentration is negligible at the entry into the steady state, then beginning with A and when k-
2
> k1, the logical expression for the initial (steady state) velocity,

The Michaelis constants of reactant A for model 1 when k-2 > k1, and for model 2 when k-3 > k1 and X and Y are in equilibrium.

*The expression for model 2 can be found from the general Km given by Haldane by approximation, using the condition that k-2 and k2 are very large. Alternatively, it is obtained from the value of y obtained when the rate equations for X and Y are equated to zero, the results are added, and x = k-2 y/k2 is substituted. Setting a = ao and b = zero, then leads to a supposed steady state value of y.
The quasi - steady states of model 2, with X and Y in equilibrium, and the correct Km
The conservation equation for E in model 2 is eo = e + x + y, and its first differential is de + dx + dy = 0. There is insufficient information to provide an exact relationship between dx and dy in the pre-steady state: it is only in the quasi- steady state itself that x, y and e each become (quasi) constant. I have therefore given an analysis of a special case of model 2, when k2 and k−2 are sufficiently large to maintain X and Y in equilibrium, a condition which allows an exact relationship between dx and dy in the pre-steady state.
In appendix i it is shown, for a reaction starting with A and with X and Y in equilibrium, that the concentration y proceeds through a maximum value only when k1 > k-
3
. The analysis essentially follows that of Morales and co-workers.4,5 I then extend the analysis to show that when k-
3
> k1, d2y/dt2 is always negative and rises monotonically to zero at equilibrium. Consequently, the only steady-state concentration available is that when y approaches the equilibrium value, yequ, and the initial (steady state) velocity would logically be written as 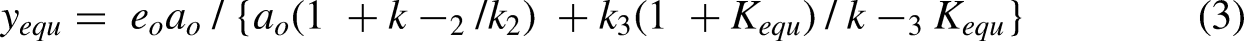


A general view of model 2
An interesting general but incomplete analysis of model 2 is obtained by comparing the equilibrium concentration of Y (equation 3) with the maximum value of y (y*), obtained from Haldane's general steady-state equation for y1,2 by setting b to zero. The condition that y* > yequ reduces to k1a
Discussion
Haldane first applied the steady state approximation of Briggs and Haldane 3 to reversible reactions in 1930, 1 and his work was republished in 1965. 2 He appears not to have considered the full requirement for a maximum, (for model 1 dx/dt = 0 and d2x/dt2 < 0, and for model 2, dy/dt = 0 and d2y/dt2 < 0), and it is probably this which led to the deficiency in his analyses. I have pointed out here that starting a reaction with A, and with k1 < k- 2 for model 1 and k1 < k-3 for model 2, dx/dt and dy/dt may approach zero only as the equilibrium concentrations of X and Y are approached: the second derivatives, d2x/dt 2 and d2y/dt2, rise monotonically to, but do not exceed, zero. In fairness to Haldane, he did state in the Introduction to the second edition of his book, 2 written shortly before his death in 1964, why it was deliberately republished unchanged. It is also possible that he was not aware of the then recent work, with model 1, of Morales and co-workers,4,5 Miller and Alberty 6 and Walter and Morales. 7
Although the final equilibrium concentration of an enzyme-substrate intermediate is, naturally, a possible solution to its rate equation, it is not revealed when, in the rate equations for models 1 and 2, the rate of change of their concentrations is simply equated to zero. The correct equations for initial rates of reaction lead to the correct analytical Michaelis constants for models 1 and 2, and these are given in Table 1.
In a steady state, the concentrations of X and Y might be in a constant ratio other than equilibrium, but an analytical solution of the rise of x and y during the pre-steady state is not possible for this condition, and it is for this reason that I have considered the special case of equilibrium (appendix i). A more general evaluation of model 2 can only be achieved by modern numerical methods. Sets of progress curves of Y could be generated, each set for a given equilibrium constant, and encompassing a reasonable range of rate constants compatible with both the equilibrium constant and the requirement that the rate constants do allow that, after the “first instant of the reaction”, the steady state is reached and b remains negligible
The condition that X and Y might be in equilibrium is not an unknown feature of multistep reactions, and has been proposed earlier based on numerical modelling (for example see. 13 This equilibrium essentially reduces model 2 to model 1, because X and Y can be considered as a single quantity. By ordinary kinetic means, when rates are measured from changes in reactant or product concentrations, X and Y are indistinguishable, and this equilibrium is one possible condition which would make sense of model 1. The latter model is a naïve kinetic view of a reaction mechanism: the intermediate X gives rise directly to two different products, A and B. Its continued use as a basis for numerical determinations of the catalytic constants 9 is only justified by the application of Ockhams razor: model 1 is the simplest to predict the observed kinetics. Haldane introduced model 2 based on experimental chemical evidence that a second intermediate may be formed after the initial binding of the reactant.
It must be concluded that, for reversible reactions, the derivation of kinetic constants in terms of rate constants must be made with caution. The application of the quasi-steady state concept requires more thought than simply equating in a rate equation dx/dt or dy/dt to zero, and if rate constants are to be back-calculated from experimental kinetic constants, the equation describing the kinetic constant must be valid. This discussion has been about single reactant-single product reactions, but it should be born in mind when multi-substrate reactions are considered.
There are other consequences of misidentified kinetic constants. The Haldane relationship,1,2 relating the kinetic constants of a reversible reaction to the equilibrium constant, is correct for models 1 and 2 only when, respectively, k1 = k-2 and k1 = k- 3 . Although Bock and Alberty 14 made well-controlled kinetic measurements using fumarase (quite reasonably assumed to follow model 1), and obtained results consistent with the Haldane relationship, it has since been shown that the enzyme is a tetramer with four identical subunits, and extensive kinetic studies by Rose 15 have shown there are several possible kinetic pathways within each subunit. Mescam et al. 13 used numerical methods to determine the relative fluxes through three possible kinetic pathways of an 11-state reversible mechanism, which they adapted from the work of Rose, and their results indicate that, except for the binding steps, each pathway had intermediate stages in a state of quasi-equilibrium. The actual mechanism of fumarase is not described by either model 1 or 2, and so whatever is concluded from the results of Bock and Alberty, 14 those results are not a confirmation of the Haldane relationship.
The kinetic constants ratio, kcat /Km, is often named the specificity constant, and this has a good intuitive basis, but it has been shown that kcat/Km is not always useful for comparing different enzymes which catalyse the reaction of a single substrate. 16 Theoretical analyses, to provide a basis for the use of kcat /Km, when a single enzyme is catalysing two competing reactions, have all depended on the use of kinetic constants deduced from rate equations by writing “dx/dt = 0”,10,11 thus failing to consider quasi-equilibria as solutions. For reversible reactions, the use of the kinetic constant ratio, kcat /Km, is only substantiated if each reactant has a binding constant greater than that of its product, or if each has a binding constant less than its product. The validity of the kinetic constant ratio as a measure of specificity therefore requires that all binding constants are known, but I am not aware of results for which this information is available.
Conclusions
Earlier work with model 14–8 has shown that, in a reaction starting with A and when the binding constant of B is greater than that of A, the quasi-steady state of X is one in which its concentration is approaching its final equilibrium concentration. I have pointed out that this leads to a correction of the equation for the initial steady-state velocity, and for the structure, written in terms of rate constants, of
As a result of these observations, I suggest caution is used if kinetic constants are derived by a back-calculation from kinetic constants, and I have concluded that the Haldane relationship1,2 (between the kinetic constants of the reactants and the equilibrium constant) is not generally valid. Furthermore, the validity of using the ratio of the kinetic constants, kcat/Km, as a specificity constant is not generally supported by the kinetic analyses10,11 of the catalysis of two reactions in a single solution by a single enzyme.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.