
Abstract
Accurate modeling of viral outbreaks in living populations and computer networks is a prominent research field. Many researchers are in search for simple and realistic models to manage preventive resources and implement effective measures against hazardous circumstances. The ongoing Covid-19 pandemic has revealed the fact about deficiencies in health resource planning of some countries having relatively high case count and death toll. A unique epidemic model incorporating stochastic processes and queuing theory is presented, which was evaluated by computer simulation using pre-processed data obtained from an urban clinic providing family health services. Covid-19 data from a local corona-center was used as the initial model parameters (e.g.
Introduction
Main objective of this work is to provide a simplified guide for building models that cover all the stages of an epidemic outbreak. Due to the discrete nature of its elements, epidemiology contains several randomness. This fact encourages us to model events of epidemiology as stochastic processes in order to increase the accuracy of the model at hand. The model presented here is not only eligible for producing reliable results, it also provides analysis of future tendencies. Another unique contribution of this paper is the formulation of the state transitions of the stages of an epidemic: (a) from healthy to susceptible, (b) from susceptible to infected, (c) from infected to quarantined, (d) from quarantined to recovered, and (e) from recovered to healthy. Furthermore, efficiency analysis of the recovery process dealing with different service/qualities is also discussed.
We do not specifically discuss the characteristics of some known approaches, for example, deterministic and stochastic approaches. We intended to design a more generic model, which can be applied to building models for human epidemics and computer epidemics with minor modifications. The approach presented here is based on stochastic processes and queueing theory. Stochasticity possessed inherently by epidemiology, and flexibility of applying stochastic models to a wide area of sciences encourages us to choose this approach for modeling epidemics on human and computer networks.
We computer engineers and medical doctor have limited knowledge of stochastic processes and queueing theory, which provide valuable methods and techniques for modeling stochastic systems. We usually describe event dynamics using models that are formalized mathematically. Although, inevitable, rigid mathematical formalization can sometimes be cumbersome and/or even disruptive when applying to experimentation and system implementation. There are, naturally, cases where complex theoretical proofs are necessary. However, there also exist areas of science that have already been discussed widely using a multitude of theories and their proofs. To obtain maximum benefit from the material presented here, we intend to simplify mathematical presentations and avoid theoretical proofs so that the models and approaches given can be easily adapted to various purposes, for example, experimentation, simulation, and resource management.
As detailed in Kondakci and Dincer, 1 lifecycle of an epidemic system generally consists of five stages, healthy, susceptible, infected, quarantined, and recovered. These are modeled as distinct states taking place in a chain of interrelated stochastic processes. The states are in most cases recurrent, omitting the case of death (an absorbing state). In any case, an infection case goes dynamically through certain transitive states, that is, moving from one state to another. Each state is numerically described either by a probabilistic function or by an integer count. The probabilistic function represents the probability of estimating the value of a given parameter, for example, probability of being infected among 100 susceptible individuals. Or, it can also describe the probability of being in a state, for example, probability of being recovered within a specific time duration. A state can also be represented by the number of increments/decrements in sizes of a parameter, for example, infected population size, the state of infectious patients undergoing quarantine operations, and the number of recovered patients.
To estimate related parameters, we generally build mathematical models fitted to some data set. Unfortunately, this is more difficult than it sounds. Because errors in statistical fitting and uncertainty in the parameter estimations are inevitable. 2 Epidemiology is often limited to the study of disease spreading in populations of humans or animals. Several commonalities between the human and computer epidemiology have recently emerged. In order to gain a general perspective, we consider that computer worms/viruses (malware) and epidemic threats to living populations (human, animal) are interchangeable. Therefore, assuming readers are from multidisciplinary environments, we avoid complex mathematical manipulations and in-depth proofs of theories, and instead focus on the applicability and correctness of the presented models. The aim is thus to provide a set of general formulae and methods that can be easily adapted to special cases in order to perform realistic trend analysis in epidemics. Simulations and experimental analysis of various cases will then be facilitated by simply modifying the general model provided.
We use stochastic approaches in conjunction with queueing theory to analyze the stages of epidemic outbreaks. Models discussed are generally substantiated by simulations and numerical examples, some of which are given here. If needed, one can refer to Taha 3 and Medhi 4 for a comprehensive coverage of Operations Research and Stochastic Models in Queueing Theory, and Cui et al. 5 for Markov models used in a multi-state repairable systems.
Motivations and related work
In–depth study of epidemiology is an extremely complex area of research. Contemporary infectious diseases evolve sporadically across a wide spectrum, for example, Covid-19 pandemics,6–8 and fundamental concepts requiring detailed study are infection spread, surveillance, treatment, control, immunity in populations, and resource management. With the Covid-19 pandemics, the world has realized the importance of the efficient resource management for health institutions, where most countries have shown significant deficiencies in their health systems. For instance, even though it has relatively efficient health system, Turkey has locked down all the polyclinics of the major hospitals throughout the country and mobilized the resources to thwart the pandemics.
So called Recurrent Epidemic Model (REM) first introduced, by Kondakci and Dincer, 1 applies a comprehensive stochastic analysis to computer epidemics, processing through a detailed analysis of a five stage stochastic model, where each stage is mathematically explored to obtain a satisfactory formalism in all dominating stages in the epidemiology. REM presents an extended structure of state transitions in order to highlight the shortcomings of the classical and deterministic models. It aims to obtain higher accuracy by use of a comprehensive model containing a sequence of stochastically dependent compartments. Other interesting approaches have been discussed in Amador and Artalejo,9,10 Yang et al., 11 and Sellke et al. 12 An epidemic model, 13 applying the finite-Markovian process gives an overview of the steady-state analysis of the evolution of an epidemic case 13 has proposed concrete solutions for determining transition dynamics by formalizing the parameters for the infection and recovery rates.
Additionally, 14 discusses the detection of worm spread, with suffeciently detailed deterministic and stochastic models. Several stochastic approaches have also been proposed to analyze dynamics of epidemics, for example, Ortega et al. 15 To gain an insight into the stochastic models, the reader can examie the survey on stochastic epidemic models presented in Britton. 16 Mathematics and simulation are effective tools assisting us in almost every field of research, in particular, for building models and substantiating the model results with regard to the theoretical background. A review of mathematical models dealing with malware propagation in computer networks is presented in del Rey. 17
Classical epidemic models (e.g. SIR, SIS, SEIR) maintain their relevance within the field of epidemic research, providing a simpler and more structural overview of epidemic outbreaks. Related to this, Artalejo and Lopez-Herrero 18 discusses the dynamics of susceptible-infective-removed (SIR) model with the aid of continuous time Markov chains. A cellular automata based SEIR model for computer virus spreading is proposed in Batista et al. 19 A stochastic approach of analysis covering SIR models is considered in Kenah and Robins. 20
Several important characteristics are common to almost all fields of research, such as mathematical biology, computer science, physics, economics, and the socail science. 21 Therefore, it is hard to confine epidemiology research to a particular field, without restricting the methods used to analyze cases that can lead to inaccurate results. For example, Mukhopadhyay and Bhattacharyya 22 discusses a comprehensive mathematical model considering the propagation of infections in human populations, which applies the theory of (traveling) waves to build the epidemic model. A comprehensive study of the mathematical epidemiology containing several models is presented in Hethcote. 23 Other interesting models can be found in Paul and Mishra, 24 Ren et al., 25 Kudo et al., 26 and Wanget al. 27
A computer worm is an intelligent virus that can automatically spread through vulnerable (or unprotected) computer applications. Some protection systems are incapable of detecting certain viruses, which are classified as polymorphic viruses, such as Stuxnet. 28 Worms are often deployed en–mass by mass–mailing in order to cause system unavailability. 29 A survey on taxonomy and characterization of computer worms is presented in Fachkha and Debbabi. 30 Numerous computer worm models exist in the literature, most of which are rooted in the classical biological models.31–34
It is also important to study the efficiency of quarantine strategies under various infection profiles. To do so, we need to have an additional stage (compartment) dealing with modeling of the quarantine process. This paper deals with a stochastic model with five compartments progressing from the healthy state to the recovered state in order to cover all the major stages in general. Effect of the incubation time on the transmission of the p. vivax malaria is discussed in Nah et al. 35
As also discussed in Kondakci, 13 many solutions assume homogeneous systems with constant transition rates, where states change with invariant probabilities. However, it has been frequently observed that both the threatened objects and their responses are heterogeneous, where states change with varying probabilities.
Communication patterns in a network is important for the propagation of viral infections. For example, the total number of bites made by mosquitoes has an important impact on the incubation time and transmission probability of malaria. 36 Immunization is naturally an important part during the course of an epidemic spreading. Accordingly, Pastor-Satorras and Vespignani 37 considers epidemics and immunization in scale–free networks applying a rigid mathematical analysis. Scale–free networks related to epidemic dynamics is considered in Fu et al. 38 Network traffic characteristics for some computer viruses may not be as important as we assume. Regarding this, Sharif et al. 39 discusses some approaches used in computer epidemics to generate propagation methods that result in successful worm outbreaks, ignoring the traffic characteristics of networks.
Models that do not incorporate the incubation period, and assume only deterministic distributions of the compartmental parameters and state changes will generally fail when fitting data to the initial outbreak data.
Susceptible nodes do not have latent periods. Since the infection cases behave differently regarding the population and infection types, the related model must accommodate the evolution of each specific case. The model presented in Kondakci and Dincer 1 discusses methods to overcome the limitations by introducing additional parameters to model the incubation periods.
For example, incubation period of computer viruses generally depends on the deployment of the individual virus and the immunity (or protection level) of the victim system. Assuming a malcode (virus, worm, trojan) embedded in an e-mail attachment, and the victims (users) of the receiving host delay opening the malcoded attachments, then the latent time might eventually be very long. Some aggressive worms like the well-known CodeRedII 40 worm, W32.Goner.A@mm, and SQL–Slammer 41 may immediately infect systems (zero latent time) without sufficient protection.
Population structures
Epidemic analysis deals with interrelated classes of populations from different networks, where the elements of each network are subject to different treatments in different stages during the course of an epidemic case. Two different types of networks are considered here, centralized and scale–free networks. Nodes in a scale–free network are connected through short paths, where the number of connections emanating from a given object obeys the power-law distribution. In epidemiology, “short path” means that the connectivity weight of two neighboring nodes is reasonably short, or the frequency of contact is sufficient to cause an infection, and therefore proper control of the epidemic spread requires more effort and resources. In contrast to scale–free networks, centralized networks have low degree of connectivity among the nodes, that is, fewer number of individuals in a population. Hence, regarding the epidemiology, nodes in a centralized networks have low rate of infection compared to the scale–free networks.
Centralized networks
Centralized networks are those that encompass many elements with low degree of connectivity. Typically, human epidemiology from small-sized communities exhibits the characteristics of centralized networks, because connectivity factors among the individuals are small and the epidemic spread is more under the control of individuals. Humans’ inherent intelligence under many circumstances can limit their access to the pathogenic environment. There are also exceptional populations who are unable to contact, for example, military wards, jails, mass production plants, complex residential areas, farms, and animal flocks. Thus, a parameter denoting the degree of contact for such networks has a significant role in the epidemiology of dense networks (e.g. intensely crowded populations).
Scale-free networks
Scale-free networks (SFN) are those that follow the properties of the power low distribution, where there is an exponential relation between the degree of connectivity of a node and the frequency of connections it makes. More on discussions about power low can be found in Newman.
42
Scale-free networks are regularly present in all forms of networks such as computer networks, social networks, and biological systems. SFNs have the property of power law, where a quantity changes as a power of another, that is,
The power low property is also associated with preferential attachment, which refers to the distribution of properties among the nodes in a network. Scale-free networks generally grow progressively by randomly joined nodes, where each node links to current hosts with preferential attachment. That is, the probability of connecting to host

For example, distribution of resources in a society will often centralize at a node with a higher degree of connections. A typical example of this is business people’s preference for commercials on highly attractive environments (e.g. TV channels, newspapers, or Web sites). Another example is the prevalence of malware attacks on highly connected computer networks in order to gain access to greater numbers of computers. Lower immunity facilitates the “invitation” of infectious diseases.
The rate of an infection a node undergoes depends on the number of contacts with infected nodes that it may have, as well as with the duration (or frequency) of the contacts. Assuming that each contact has the same infection rate denoted by

where
Growth and extinction dynamics
Let us consider the classical SIR (Kermack–McKendrick model
32
) model and expand it to contain the dynamics of healthy

where
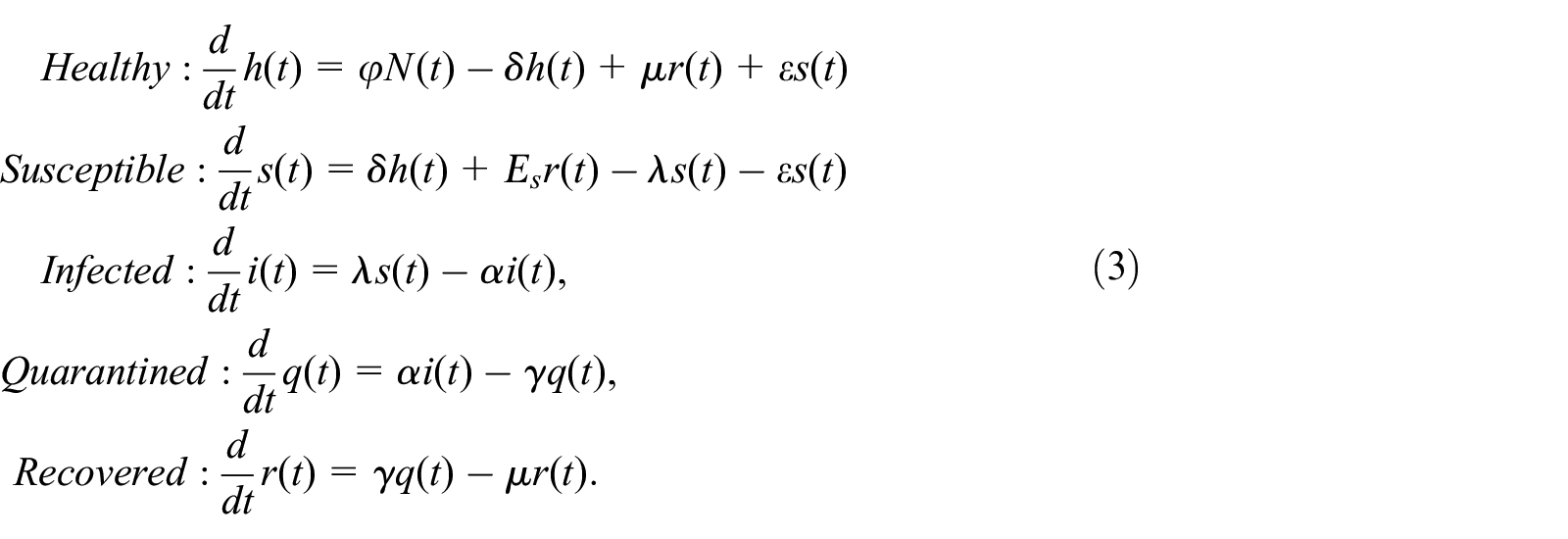
The parameters are self explanatory, for example, population change in a compartment is interrelated with the parameters of its predecessor and ancestor compartments. For example at time
A distinct property of the ESIR model is that a very small proportion,
Basic reproduction number
The concept of basic regeneration number was first used in conjunction with demography in 1880s to predict the growth rate in specific sections of populations.
45
Regarding the epidemiology, it expresses the number of potential transmissions from a single infected node during its transmitter period, and is denoted by

Where,
There exist numerous methods to define

Where,
According to World Health Organization (WHO), Vector-borne infections make up 17% of all infectious diseases; these are transmitted by mosquitoes, triatomine bugs, blackflies, tsetse flies, sandflies, mites, ticks, lice, and snails. Hence, finding an appropriate formula of

Where,
Epidemic threshold
This is conditional on the fact that prior to reaching an epidemic outbreak, there should be a large number of susceptible hosts exposed to infection, regardless of the probability of an initial outbreak. An epidemic threshold can be reached when at least one infectious host is in contact with susceptible hosts for a specific duration. Branching process is a useful approach for modeling many physical events, as well as epidemic outbreaks. An initial epidemic outbreak often evolves as a branching process in a crowded population without preventive control. For example, infection has a slower changing stochastic behavior in a human population compared to poultry. On the other hand, unprotected computer networks have a rapid evolving of infection characterized as a typical branching process. In most cases, effective infectious periods for computer networks are brief, whereas these periods may last a lifetime for human populations. Predicting the epidemic threshold and the basic reproductive number in such cases can be extremely complicated, if a certain threshold of observation data is not available.
The probability of a single infection producing a new infected node with rate

whereas the probability of a node without infection is expressed by


Now, let

be the probability of recovering from each disease with rate

Thus, the probabilities for zero and

where

The probability of zero production,

Rather than the notion of the basic reproductive number
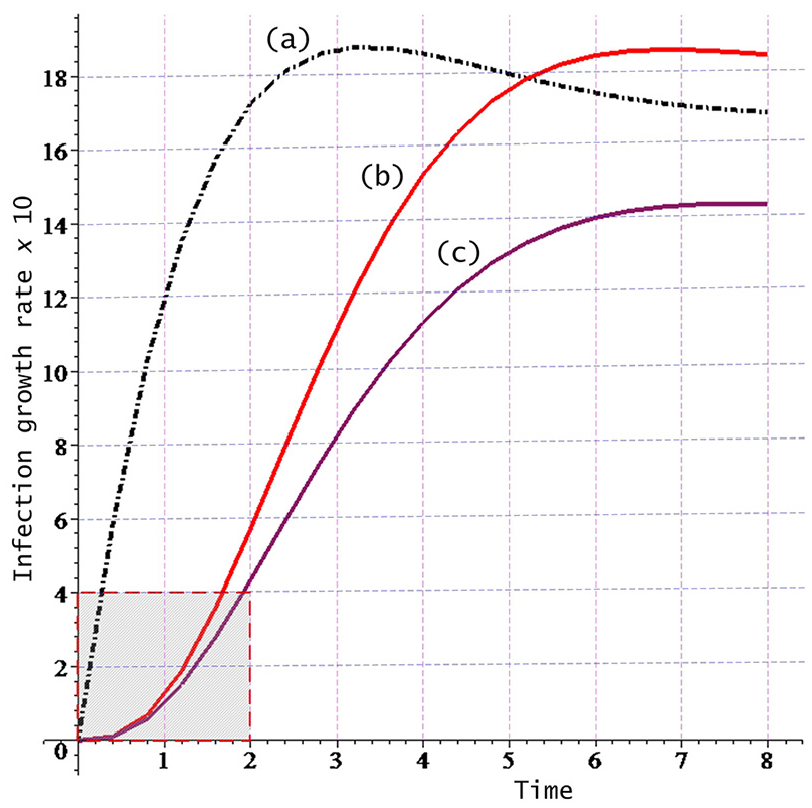
Infection growth rates for different epidemic threshold values: (a) Covid-19 initial spread rate in Turkey, (b and c) different forms of typical influenza.
Stages in epidemic cases
Spreading and recovery operations i a typical epidemic case follow a set of contiguous and interdependent stages, where each stage is modeled as a dynamic subsystem with adequately chosen state parameters. Each subsystem has some input and output parameters which change dynamically throughout the lifecycle of the epidemic case. In most cases, the subsystems are transient, and each subsystem feeds the input of its ancestor subsystem with data, while receiving data from its predecessor subsystem. Data exchanges between the subsystems take place through the individual state parameters of the associated subsystem. Each subsystem has its own population of elements, where the number of elements in a subsystem is denoted by a numerical value called “state.” For example, state
Compartmental states associated rate parameters.


A compartmental representation of the states of the recurrent model.
As can be noted, elements of the populations iterate from state to state depicting a recurrent characteristic, where the population sizes are precisely expressed by the following state equations, equation (8).

As also illustrated in Figure 2, some fractions of elements are flowed from population to population at different intensities, for example, from healthy to susceptible at rate
State variables contain the time-dependent number of objects in a given epoch
Using notations from queuing theory, the stages (compartments) of the generic model will be described in the following sections. Arrivals of an object to a stage and operation completion (departure) from a stage are modeled as an M/M/1:(FCFS) queueing system. Based on this first-come first-served (FCFS) principle, each stage in the system encompasses a cascaded queueing system, where each stage serves a unique function. The overall behavior of the system is illustrated by the flow diagram shown in Figure 2, where each stage is a dynamic component of the recurrent operations. Initially, at least one host must be infected in the Susceptible population for further development of a disease. Often a stochastic growth of the susceptibility takes place as a branching process. Thereafter, an epidemic outbreak (exponentially distributed process) is initiated in the susceptible population. Arrivals to the infection stage and most other stages are modeled as a Poisson process denoted as
A generic model
In order to facilitate the analysis in a broader scope, a generic model is necessary, which can elucidate the most encountered stages of epidemiology. In order to support this idea, the model presented here is built upon five types of populations: (i) healthy, (ii) susceptible, (iii) infected, (iv) quarantined, and (v) recovered. Furthermore, we have two major assumptions: (a) all individuals/nodes in a sample population are initially susceptible, (b) if a node gets infected then it becomes the source (transmitter) of a branching process for spreading the infection. At the next time slot, some of the hosts in the population are susceptible, and some are transmitter, which can cause infections. Initialization of the susceptible population with empirically obtained data is necessary in order to keep analysis within an acceptable dynamic range until the equilibrium state is achieved.
Unless otherwise specified, the process of infection through recovery is considered as an M/M/1 queueing system with a single server having Poisson arrival (infection case) and exponentially distributed recovery service time (recovery case). More specifically, the system is an FCFS Markovian queue with exponentially distributed interarrival and departure (or service) rates: arrivals and departures are independently and exponentially distributed with rates
Two distinct populations (biological and computer) may have different levels of susceptibility, where the susceptibility of an object depends on the protection level for computers and immunity level for living individuals. The immunity level for living individuals has several parameters such as age, locality (e.g. kindergarten, chicken poultry), mobility, vaccination, and so on.
Spread and extinction dynamics of a disease can be analyzed by two fundamental methods, time-dependent and limiting (or equilibrium) behavior analyses. Analysis of time-dependent behavior can help determine the number of patients (or hosts) under care by computing the probability that at time
Definitions
Healthy to susceptible
All starts with a susceptible object having contact with one or more transmitter objects. The susceptible objects can be infected and become an active transmitter after the latent period. Following this, an epidemic outbreak will occur. The outbreak is best modeled as a branching process, see Athreya and Ney 57 and Kalinkin 58 for a general review of branching processes. A branching process is a regenerative Markov process, where an individual in a population goes through a recursive regeneration process that proliferates in the next generation with a specific probability.
Let us assume a group of transmitter objects have contact with some susceptible objects, where the contacts and eventual infections are independent of each other. Here, the probability of a contact causing

Besides these one-step transitions, transitions from
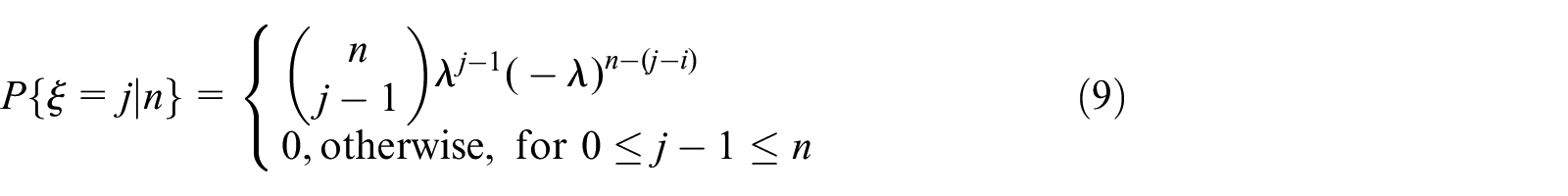
The Poisson approximation of the above distribution is

A small proportion of the susceptible population may be well-protected or may have sufficient level of immunity, and hence, will not become infected. Hence, the proportion of the susceptible hosts returning to the healthy population is depicted by

Susceptible to infection
Assume that we have a population consisting of

However, if the population has a small number of individuals, in which each infection being considered as a “success” in a Bernoulli process. Then,
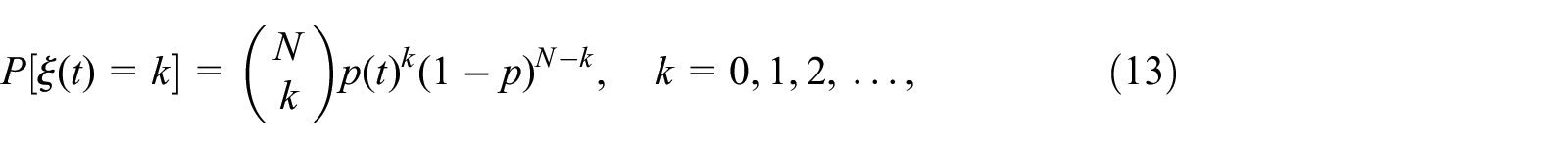
As can be noted, the infection phase consists of two sub–phases: first, some susceptible hosts, which are vulnerable (or unprotected), will be infected with a rate depending on the infection probability and density of the susceptible nodes. A second sub-phase consisting of a virus spread that starts through the currently infected nodes, as a branching process expressed by the following distribution, 1

where
Example
Assume we have a record of 180 infection cases a month. We need to determine (a) the probability of eight incidents per day, (b) the probability of at most five cases in a day
(a)There are

(b)
The results of a simulation with the Poisson distributed infection threshold and branching convoluted with Gamma distribution are plotted in Figure 3.

Illustration of rates for the spread of infection evolving as a branching process.
As can be noted, recovery starts soon after the infection starts. This enforces a small diminish in branching rates modeled as Gamma function. Gamma distribution is tied with the immunity growth that enforces annihilation of the spread, with the degree of immunity. The curve in Figure 3(b) shows the branching rate changes with an insignificant incubation time, whereas 3(c) shows the effect of latent incubation times.
Infection to quarantine
Assume that, following an epidemic outbreak, we have a group of arrivals of patients with a Poisson rate

a single discovery in the time interval

the probability for more than one cases is expressed by

The diagnose process proceeds as follows. It will be shown that

The infection–to–quarantine rate parameter,
Let us now determine the distribution of the transition time from infection to the quarantine stage, which we model as a memoryless process, where let the state

where

will obtain the probability of event

and hence,

Then, the random variable


Thus, the infection to quarantine time is exponentially distributed.
Quarantine to recovery
As shown in the previous sub-section, nodes in the infected population (or network) are inspected, where false positives are discovered in significantly short times. Regarding the computer viruses, users generally discover malfunctioning of computers at random when they turn on the computer. A simple birth–death process is appropriate for modeling the discrimination of real infection events and false positives, where a successful diagnose of an infection and a false positive can be obtained by

Here,
For the stationary state, the probability of exactly

where,

The average number of nodes discovered as infected and awaiting for recovery is given by

Mortality rate
The probability

as the average number of infected objects, whereas the number of lost objects is simply given by

The reader is encouraged to determine the mortality in terms of
Recovery to healthy
Total recovery (or hosting) time for individuals is denoted by an exponentially distributed random variable,

will give the probability for determining the total time required for the overall hosting of a patient. Let the times between various hostings be denoted by a Poisson process,

Hence, the mean time between recoveries is given by

The recovery process described by equation (25) can be simply modeled with two states, idle
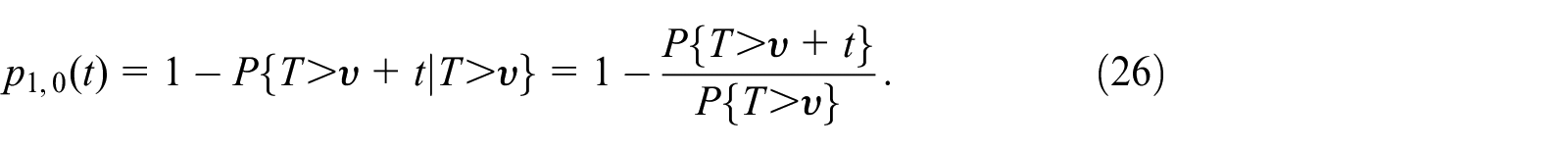
Hence, plugging equation (25) into equation (26), we get the simplified equation for the transition probability from busy to idle state as

Obviously, we have a Markov process satisfying the following conditions

We have the facts

and

to our disposal. Transition density from state

Recovery facility with single serviceman
We need to determine the limiting solutions so that we can easily determine the mean requests awaiting for the recovery at any time. Suppose we have a service facility with a single repairman (server) handling at most

We can obtain the limiting probabilities by solving the following system of equations

Hence, solving this system of equations, the limiting probability of exactly

Thus, based on the condition

We can now determine the limiting probability of exactly

Thus, the limiting probability

It should be noted that these analyses assume
Example: Resource utilization
Let us assume that a healthcare center randomly receives an average of 128 patients each 24 h. The center spends on average 8 min on each patient, where each individual is treated one at a time. Based on this information, we can determine a set of useful results regarding (a) proportion of the time the center is busy, (b) system utilization, and (c) mean service rate, (d) probability of the number of patients at the center, (f) idle time of the system.
Assume that the care center is modeled as an

Average service rate per hour is

System utilization (proportion of busy time) is

Probability of having the system idle (i.e. exactly 0 patient under care) is

Probability of having exactly one patient under treatment is

Probability of having more than one patient under treatment is

Recovery facility with random service rates
Throughput of a service facility is one of the dominating factors for jobs with large and randomly fluctuating backlogs. It is also important for us to introduce this case here. Generally, higher workload may lead to a situation of “panicking” server resulting in diminished throughput, whereas lower workload may divert system to a “lazy” server, also resulting in diminished throughput. It is often suitable to assume constant mean service rates for systems involving automated or machinated services. However, in reality, queueing systems involving human servers cannot always have constant rates. For example, increased incoming requests (e.g. patient flow into an emergency unit in a hospital) may result in increased pressure on the doctors/nurses undertaking the service. This will, in turn, result in increased effort and higher service rate, or even cause a worsened situation due to the higher pressure. On the other hand, when the backlog (queue length) is small, then the service rate will decrease, due to the “lazy” server syndrome. More on the related issues can be found in Gross et al., 59 Stewart, 60 and Asmussen. 61 One can also refer to Schwarz 62 for more detailed analysis of time-dependent queueing systems.
Suppose we have a system with state space

need to satisfy the forward Kolmogorov equations

We then have for a system having stationary characteristics

Hence, we have the limiting distribution

where

On the basis of the non-catastrophic constrain we also have the following relation.

Again, assuming Poisson arrivals with

where,

The negative effect of large backlogs leads to decrease in the service rate. This can be due to the annoyance of individuals waiting longer than expected, or diverting the requests to other facilities, if available. Such cases will typically lead to decreased arrivals, where a random coefficient,

This random fluctuating with large backlogs is illustrated in Figure 4, which depicts the result of a simulation with random mean service rates satisfying the relation of

The effect of fluctuating arrivals causing increased service times: (a) input queue, (b) increased service time with random fluctuations affected by large backlog, and (c) decreased steady-state departure rime.
Experiments and simulations
One may need to experiment a chosen model with different cases and scenarios in order to validate it, that is, to determine whether it can produce expected results. Experiments should be better combined with simulations so that they can be numerically verified based on various scenarios. It is usually trivial to simulate computer epidemics due to more concrete definition and fewer number of the system parameters compared to that of human epidemics. In fact, some human viral infections behave similarly to that of computer infections, and can be easily adapted to computer simulations. For example, some human/animal viruses attack certain cells and proliferate within the attacked victims in the same way as a computer worm spreads itself. Therefore, we can modify some validated computer virus scenarios to match the biological viral infection cases and scenarios. The scenarios can then be experimented with in a network laboratory to observe the spread and control of infections, and the results are then justified with computer simulations. The simulation can then be expanded to evaluate a variety of biological epidemics, if desired. For example, CMV: cytomegalovirus, HPV: Human Papilloma, HTLV: Human T-Lymphotropic, rubella, influenza, Herpes 1 and Herpes 2 are the family of human viruses, where their propagation patterns can be justified by computer simulations, if appropriate models with sufficient amount of data are available.
In order to emphasize the diversity (or similarity) between the human and computer epidemics, a simulation of two related cases was conducted, where both processes have Poisson input, but their recovery processes evolved differently. It was shown that the human recovery process involves the Gamma distributed recovery process. This result is realistic, due to the automatically grown immunity found in human intelligence.
63
Figure 5 illustrates results of the simulations with constant infection rate of

Simulation results depicting the infection and recovery characteristics of human and computer cases each having 1000 hosts.
The recovery is exponentially convolved with Gamma distribution. Gamma convolution increases the accuracy of recovery due to increased immunity of the infected objects while undergoing recovery. Based on a simulation, the changes in a population of 1000 nodes are shown in Figure 5.
Treatment evaluation
The efficiency of a healing method (or protection) can be evaluated to determine whether it is reliable during the course of its application. The model given here is straightforward, where the probability of a single failure and success of a treatment develops as a simple birth–death Markov chain. It is assumed that healing fails with rate
Hence, we can deduce the following set of equations to describe the probability of state transitions occurring during

These linear equations deal with a single item, which can be in any of two mutually exclusive Markovian states at a time, either failure (birth) or success (death). Hence, failure of a treatment is depicted by a birth process, whereas a success is depicted by a death process. Prior to solving these equations we rewrite them in the form of differential equations as.

Solutions of these equations can be obtained by applying various methods, for example, Laplace transform, recursive numerical method, Langranges, and transition matrix. Thus, by setting


Predator–prey cases
In computer networks, we can have predator software that can fight against malware in real-time. Internet pop-up and Trojan blockers should work in this way. However, most anti-virus programs are usually configured to run periodically or manually, which then cannot be considered as dynamic predators. Because computer malware (worm, virus) are frequently and randomly deployed in different forms, discovering them in real-time becomes extremely difficult. The predator-prey model has a periodic behavior because the effect of hunting and surviving changes relative to the respective population growth rates. Lotka-Volterra predator-prey model has been frequently referred to for describing the relation between a predator and its prey, where the predator consumes the prey to survive. Increasing the level of consumption leads to higher starvation rates, especially if a certain prey is the only source of survival for many different predator types.
Let us simulate a scenario dealing with computer viruses in order to observe the power of predators behaving as a benign computer virus. The following differential equation, equation (42), models an anti-virus as the prey, where a computer virus plays the role of the predator, which is also programmed to destroy (stop) as many as anti-viruses as possible. Population parameters are

Figure 6 illustrates the simulation results obtained from the model given in equation (42).
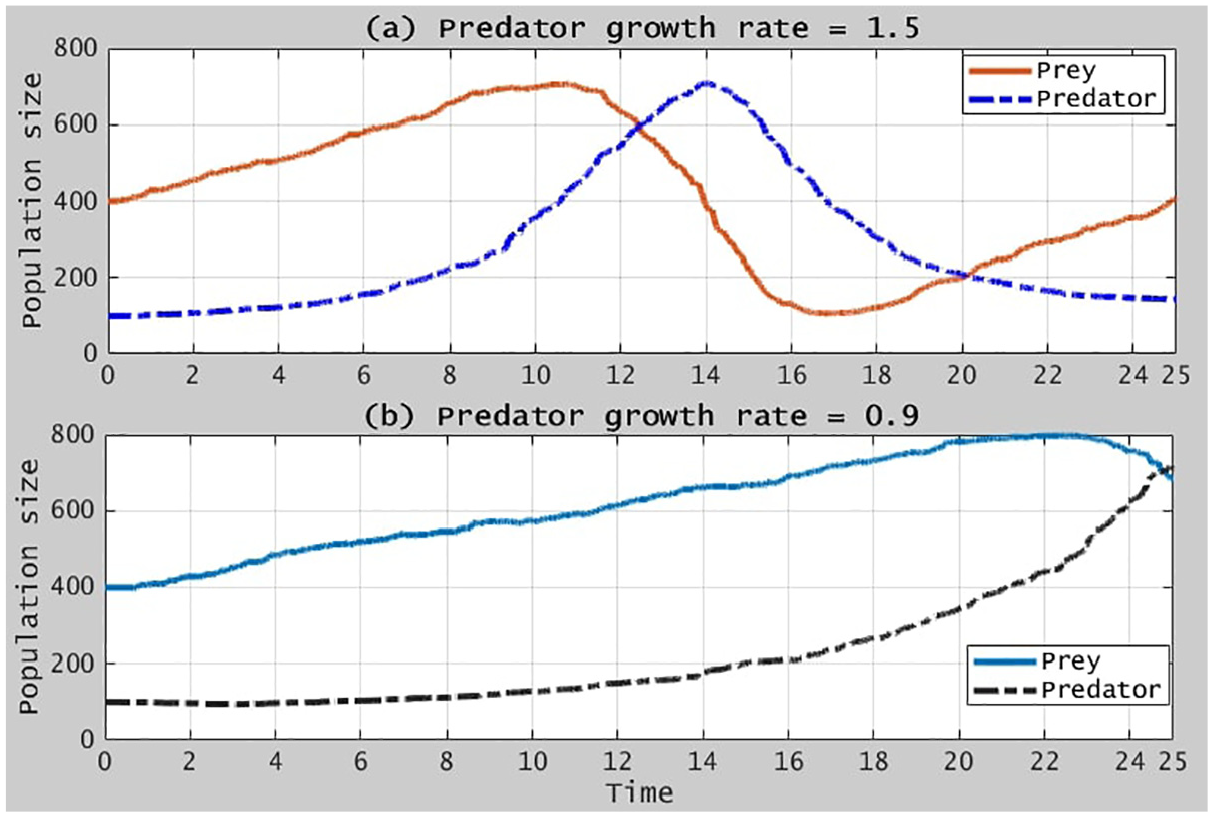
Illustration of the predator-prey relation.
As shown in the figure, the rate changes have also random characteristics, because virus attacks are random, that is, by nature hunting and surviving occur in a random fashion. Ideally, the system can come to an equilibrium point, where the effect of infection rate will be governed by the prey population as shown in Figure 7.

Illustration of the steady–state in the predator-prey extinction state.
Discussion
Designing accurate models can significantly mitigate the number of cases and mortalities. Models developed for computer epidemics cannot be directly applied to human epidemics, and vice versa. Computer epidemics differ from human epidemics in some aspects. For example, incubation (latent) time for computer viruses can be extremely short, and even in some cases unpredictably long. On the other hand, latent time depends generally on the type of virus for human epidemics, and depends minimally on the individual’s immunity.
We generally consider analysis of epidemics under two different numerical approaches, deterministic, and stochastic. Deterministic models can be simple to apply, in which the limiting distributions can be obtained more easily with fewer parameters. On the other hand, stochastic models have more sophisticated stages of analysis, which allow fine-graining of parameters for more precise results. This feature leads naturally to more time needed for analysis in order to achieve the equilibrium state, hence manual computation of results is infeasible for large data set, for example, more than 10 observations. Another limitation is the level of learning threshold, which may hinder its application in some environments.
Designing of an extended stochastic model is planned as a future work, in which the extension will deal with new births (connections coming from external networks), mortality, and cases of pandemics dealing with infectious agents exported to external networks.
Conclusions
It is important to build models reflecting realistic characteristics of a given case, including effective resource planning, estimation of future risks, and reliable measures for mitigation. This paper presented a generic model for analyzing the epidemic stages and their interaction with each other. In most cases, epidemiology is a stochastic process involving several stages of evolution. Therefore, applying queueing theory enhanced with stochastic analysis can give a better overview over the dynamics of epidemic cases. Although the correctness of other models (e.g. deterministic, diffusion, differential analysis) is undeniable, features of stochastic queue models can guide us to simpler and more structured solutions that can be realized using recursive computer algorithms and other numerical approaches. This paper has modeled all stages of an epidemic case, where each stage has been formulated thoroughly within a stochastic context, so that the approach can serve as a guide for building models applicable to different environments with stochastic behavior.
The approaches given here are applicable to both human and computer epidemics. Test and verification of models used for human epidemics can be validated by an associated computer model, where computes can be used as test objects representing the individuals of a human population. By this manner, we can acquire some key parameters that can be precisely used for the human model. For example, parameters such as immunity, incubation time, and infection rate can be adjusted to match the characteristics of a given virus attacking humans, and then run simulation with computers to obtain results reflecting the human epidemics caused by that virus. In order to increase the accuracy of a model we can set up various infection sceneries and configure a wide range of parameters and network sizes/types in a computer simulation. Hence, we can precisely obtain the characteristics of a given type of viral outbreak in a human population by fine-adjusting of the variables used for the simulation with computers as victims.
Footnotes
Appendix
Appendix B: Data sampling and rate estimation
Assuming a Poisson process with a collection of random observations that are independent and identically distributed, where the random variables all are mutually independent and have same probability distribution. If we want to determine an estimator for the rate parameter
In order to facilitate the computation of the estimator, the corresponding log-likelihood function of the joint density function followed by a derivation can be applied to find an estimate of the unknown parameter
and taking the derivative of the likelihood equation, equation (60), leads to the maximum likelihood estimator for
Acknowledgements
We would like to express our deepest appreciation to reviewers of this paper for their constructive comments, and Simon Edward Mumford for his excellent contribution in proof–reading of the paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.