
Abstract
Mycosporine-like amino acids have long been known as a natural form of photoprotection for fungi and cyanobacteria. This review will highlight the key time-resolved experimental and theoretical techniques unravelling their photochemistry and photophysics, and directly link this to their use in commercial skin-care products, namely as sunscreen filters. Three case studies have been selected, each having aided advancement in this burgeoning field of research. We discuss these studies in the context of photoprotection and conclude by evaluating the necessary future steps towards translating the photochemistry and photophysics insight of these nature derived sunscreen filters to commercial application.
Keywords
Mycosporine and mycosporine-like amino acids
Ultraviolet radiation (UVR) can be both beneficial and deleterious to life.1–4 To combat the detrimental effects, nature has evolved to include several collections of UVR absorbing compounds, each biologically engineered to protect their host, while allowing sufficient absorption in different regions of the electromagnetic spectrum for important biological processes.5–8 The recent controversy surrounding artificial sunscreen filters focuses largely on their potential to act as endocrine disruptors and skin irritants to humans.9–12 Alongside these, there are growing concerns surrounding the environmental impact sunscreens filters are having; for example, the common ultraviolet (UV) sunscreen filter oxybenzone has been linked to coral bleaching and reproductive toxicity to aquatic life.9,13–15 These findings have therefore fuelled research into alternative filters, such as those derived from nature.16,17 One such class of natural UVR absorbers is that of mycosporine/mycosporine-like amino acids and their derivatives (both termed MAAs hereon). (Mycosporine refers to systems based on the cyclohexenone core, and mycosporine-like amino acid refers to those based on a cyclohexenimine core. See Gao and Garcia-Pichel 18 for further details.) MAAs are a common form of photoprotectant species, found in fungi, cyanobacteria and phytoplankton. This review focuses on their photochemical and photophysical (collectively termed photochemical hereon) properties and how this knowledge can be utilised to develop next generation sunscreen filters. Importantly, the reader is directed to the following excellent reviews for further details regarding MAA biosynthesis and extraction, areas that have already received considerable attention.7,8,18–23
MAAs present a promising form of future sunscreen filter given their strong UVR absorption, with typical extinction coefficients in the region of 30,000 M−1cm−1. 8 Currently, approximately 30 MAAs have been isolated which, combined, cover the UV region (UVA = 400–315 nm, UVB = 315–280 nm and (in part) UVC =280–100 nm) of the electromagnetic spectrum.7,8,24 Derived from a basic cyclohexenone or cyclohexenimine core, Figure 1(a), each having a differing absorption maximum, these properties lend themselves towards use in future cosmeceutical applications, largely due to the ability to choose the range over which particular MAAs absorb. This variability in absorption is achieved through selective biosynthesis of MAAs with differing functional groups onto either position 1 or 3 of the core structure (Figure 1(a)). 25 Substitutions far removed from these two positions have little effect unless a large change in conjugation is encountered, resulting in a change to the overall chromophore (highlighted in red in Figure 1(a)). 18 The exploration of differing functionalisation on positions 1 and 3 not only allows for variability in the electronic ground state absorption spectrum of the MAA, but is necessary for widespread commercialisation as natural source extraction and synthetic preparation yields only small quantities of natural MAAs (typically 15 steps, 1% of overall yield).8,26–28
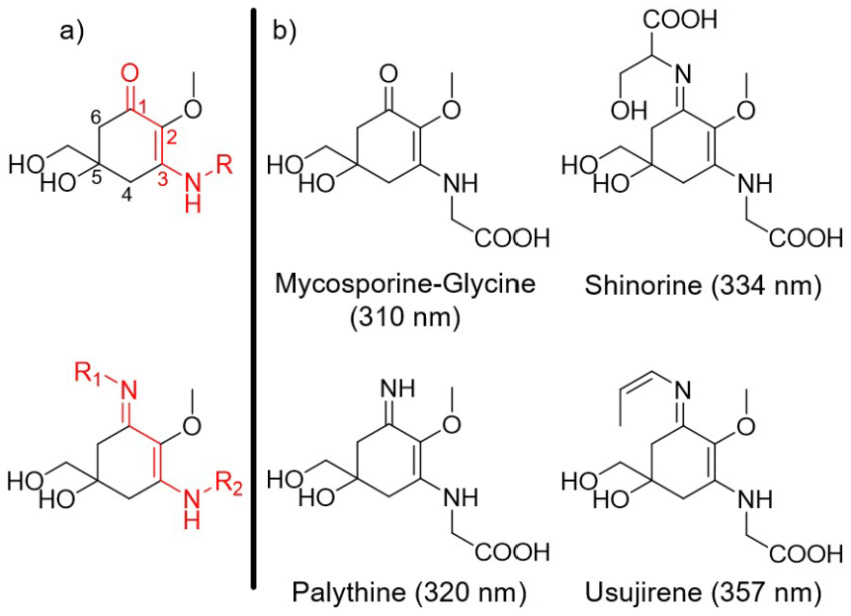
(a) Core structures of mycosporine (top) and mycosporine-like amino acids (bottom), with the chromophore highlighted in red. (b) Common mycosporine and mycosporine-like amino acid structures along with their reported absorption maxima adapted from Bandaranayake. 8
Sunscreen filters work by facilitating the safe dissipation of absorbed, potentially harmful, UVR as heat to the surrounding environment. 29 This review will highlight the main experimental, and to a lesser extent, complementary theoretical techniques used to track molecule-photon interactions (photochemistry), and hence unravel the photoprotection mechanisms underpinning MAAs. This, in turn yields important insight into why nature has chosen these particular sunscreen filters as photoprotective species. Furthermore, we will briefly discuss how we can adapt these techniques to accommodate a truer cosmeceutical environment. This review will then discuss three key articles – termed Case Studies – which put into practise these experimental and theoretical techniques. Using findings from natural MAAs and other sources,16,17 ‘rules’ for efficient molecular design can be established in relation to increasing the efficacy of next generation sunscreen filters. This review closes by discussing future steps towards translating photochemical insight into application.
Tracking the energy flow within molecules
There are many articles and reviews in the literature that discuss methods to track population flow within a molecule’s excited states, along with their advantages and disadvantages. The reader is therefore referred to the following excellent sources for such detailed discussion.30–35 What follows is a brief overview of the commonly used experimental and theoretical techniques used to track the evolution of photoexcited states pertinent to the Case Studies to be discussed below.
Transient electronic absorption spectroscopy
Time-resolved pump-probe spectroscopic techniques such as transient electronic absorption spectroscopy provide powerful insight into the fate of photoexcited molecules.34,36 An initial femtosecond (fs; where 1 fs = 1 × 10−15 s) laser pulse within the UVA/UVB region photoexcites, or pumps, the sample of interest to an electronically excited state. This photoprepared excited state is then interrogated with a second laser pulse termed the ‘probe’, typically a white light supercontinuum spanning wavelengths between 300 and 700 nm. 37 The relative time-delay (Δt) between the pump and probe is varied, usually, from femtoseconds to nanoseconds (ns; where 1 ns = 1 × 10−9 s). Changes in optical density (ΔOD) are calculated between the probe, I0 (λ, t) traversing a photoexcited (via interaction by the pump) and unperturbed sample I0 (λ).
A schematic of a typical transient electronic absorption spectroscopy system is displayed in Figure 2, presented as three key sections. First, in Figure 2(a), ultrafast pulse generation is commonly produced through the use of a Ti:sapphire oscillator combined with regenerative amplification to generate an 800 nm fundamental fs pulse train with a pulse energy of a few mJ,38,39 and a pulse duration of 10 s of fs; these levels of output energy allow for the generation of both the pump and probe pulses through nonlinear optical effects.37,40,41 Second, in Figure 2(b), the output of the laser system is split to generate the pump and probe pulses. Probe pulses follow an optical delay line (in this example) that includes mirrors and a retroreflector mounted on a delay stage, which enables Δt to be varied (by virtue of increasing/decreasing the optical path length of the probe) before focussing the 800 nm beam onto a calcium fluoride (CaF2) window to generate the aforementioned broadband supercontinuum. 40 The probe pulses are then focussed into the solution (within a liquid flow cell, see below) containing the solute under investigation. Finally, in Figure 2(c), the transmitted probe is collimated (not explicitly shown) and sent into a spectrometer.
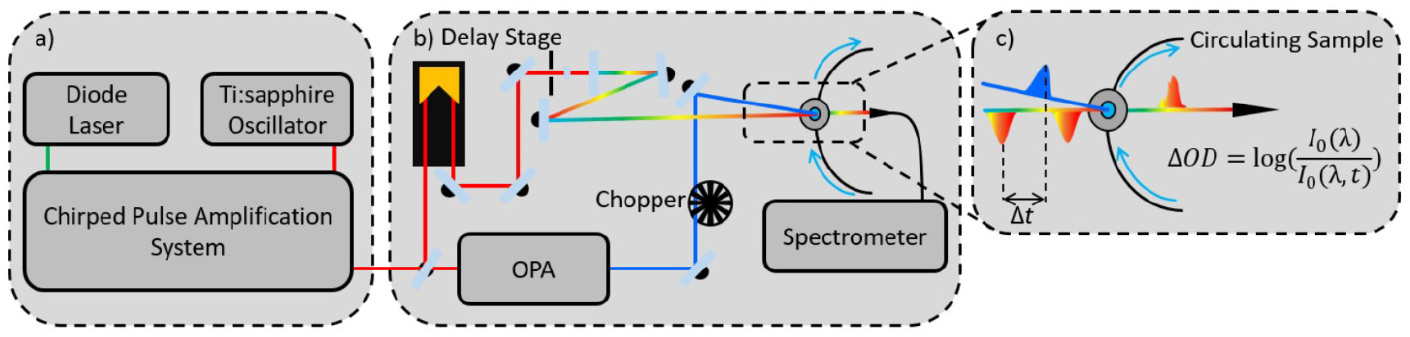
Key stages of a transient electronic absorption spectroscopy system: (a) ultrafast pulse generation, (b) pump-probe setup and (c) sample interaction and signal recovery.
Pump pulses are typically generated through optical parametric amplification of the 800 nm beam, which allows for the variability of pump wavelengths across the UV spectrum (in the present Case Studies UVA/UVB) and beyond. The pump beam is spatially overlapped with the probe beam within a liquid flow cell containing the sample solution. The solution is recirculated which enables fresh sample to be interrogated for each measurement. Importantly, a chopper wheel blocks every second pump pulse to allow for calculations of the change in optical density (ΔOD; see Figure 2(c) for details) between pulse pairs (i.e. pump ‘on’ and pump ‘off’). The collected data, termed transient absorption spectra (TAS), are then commonly displayed as false colour heat maps (vide infra). These two-dimensional plots show the full probe continuum as a function of pump-probe time-delay, with the change in optical density shown on the z scale as changes in colour.
Photoacoustic calorimetry
Photothermal techniques such as photoacoustic calorimetry provide quantitative information on the energy flow between solute and solvent environment.31–33 Photoacoustic calorimetry allows for precise thermodynamic properties and lifetimes of a photochemical reaction to be derived through monitoring of the pressure changes induced through photoexcitation. 42 These pressure changes, depicted by orange bars in Figure 3, are initiated usually by ns pulsed UV lasers, depicted by the green bar in Figure 3, which traverse the solvated sample held in a cuvette at a given temperature. The transducer monitors the pressure change commonly in a right angled orientation or alternatively a forward facing orientation, in which case a dichroic mirror is placed between the sample and transducer; these are shown, respectively, by Figure 3(a) and (b).31,33 A typical photoacoustic signal is displayed in Figure 3(c), this signal is formed from two contributions: (1) the thermal expansion of the solute upon the heat being delivered through radiationless relaxation and (2) the volume change induced in the solute and solvent.28,29 Through performing the experiment at multiple temperatures and comparing with a known calorimetric standard, the initial peak to peak amplitude can be assigned as the energy lost from the solute, H in Figure 3(c).28,29 A calorimetric standard is usually a compound that presents a similar absorption and undergoes no radiative transitions, that is, fluorescence or phosphorescence. 43
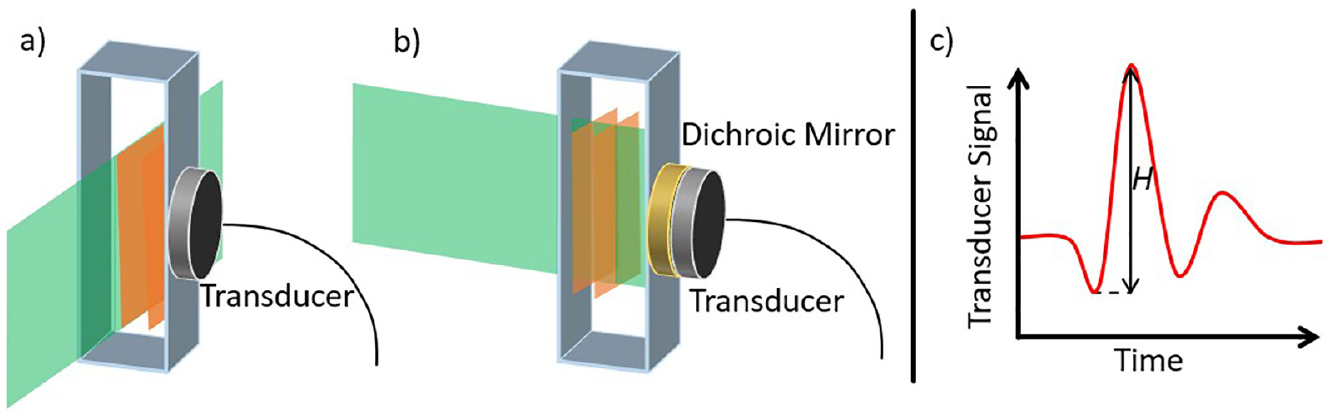
Sample interaction for photoacoustic calorimetry with (a) right angled and (b) front facing geometry. Incoming laser pulse is shown in green, with induced pressure changes shown in orange. These changes are detected by a transducer connected to an oscilloscope. (c) Expected signal from a photoacoustic calorimetry measurement showing the initial amplitude H.
Theoretical methods
Computational exploration of the ground and excited electronic states of molecules can allow for complementary insight into the molecular processes undergone in molecules following photoexcitation, some of which may be undetectable utilising the experimental techniques discussed supra. Electronic state calculations for the electronic ground states can be considered routine for numerous systems, given a single electronic configuration. 30 Challenges for electronically excited states often arise from the need to consider numerous electronic configurations, as well as different ranged interactions, such as those discussed below. Intermolecular interactions (such as hydrogen bonding, clustering and aggregation) can also alter the excited electronic states of systems further to the intramolecular considerations which one encounters in vacuo. Polarizable continuum and conductor like screening models can be applied to account for the electrostatic nature of the solvent interaction.44,45 However, to fully capture the explicit nature of solvent interaction and systems such as those described above, particularly hydrogen bonding, for example, multiple solvation shells are regularly required along with one of the aforementioned models to achieve suitable levels of accuracy.46–48
Ab initio methods make use of self-consistent field (SCF) methods and Hartree–Fock Slater determinants to define a full wavefunction for the system using a set of given orbitals and a prescribed number of electrons. A particular approach, known as complete active space self-consistent field (CASSCF), has advantages in finding degeneracies between excited states, so-called conical intersections (CI), which often drive non-radiative relaxation of the excited state population (crucial for an effective sunscreen filter, as will be discussed later).49,50 The disadvantage of such a method is the computational cost associated with such calculations (CASSCF is also commonly employed with a Møller–Plesset perturbation (CASPT2) to aid the accuracy of calculation; however, this does however increase the computational costs). To remedy this, such calculations often only involve key orbitals and electrons (often those located within the light absorbing ‘chromophoric’ component of the molecule); reducing the number of orbitals and electrons considered reduces the quality of the result.30,51,52 Furthermore, additional techniques such as Time Dependent Density Functional Theory (TDDFT) which works analogues to density functional theory, by applying functionals to model the spatially dependent electronic density of the molecule under study, are regularly used to reduce these computational costs for vertical excitations and excited state geometries; 53 however, TDDFT is unable to locate CIs.
Case studies
In the ‘Tracking the energy flow within molecules’ section, we have discussed three key techniques which allow for insight into the excited state dynamics of systems of interest herein. We will now highlight three key papers (along with associated works), pertaining to the excited state dynamics of MAAs, which have each represented an important step forward in the photochemistry and photophysics of natural sunscreen filter research. We note that these Case Studies display a small, but we hope representative, selection of this burgeoning area of research. Case studies 1 and 2 detail both theoretical and experimental excited state lifetimes of MAA-inspired systems. Case study 3 discusses how energy flows within a solution from the photoexcited solute to the surrounding solvent environment.
Case study 1
One of the major challenges towards commercialisation of MAAs is the lack of their simple organic synthesis. While protocols have been reported for their synthesis or extraction, most require multiple steps and have low percentage yields.26,28 Work by Losantos et al. 27 has explored the functionalisation of the core moieties of MAAs (shown in Figure 4) and their consequent excited state dynamics properties theoretically, through the use of CASSCF methodology (see section ‘Theoretical methods’), in an effort to mimic the natural (MAA) systems. Importantly, this reduces the complexity of organic synthesis, as these MAA-inspired systems require fewer synthetic steps along with a greater percentage yield. 27
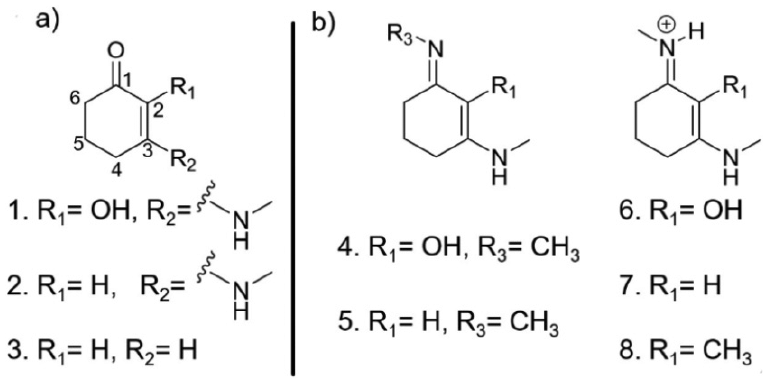
Chemical structures of systems studied by Losantos et al.: (a) cyclohexenone core structures and (b) cyclohexenimine core structures.
Employing a computational approach, Losantos et al. extracted key points along the minimum energy pathway (CIs and potential energy surface minima) for the core structures with differing functional groups, based upon the three cyclohexenone and five imine core structures given in Figure 4. Their findings show that MAA-inspired moieties that possess a cyclohexenone core prevent repopulation of the electronic ground state; following photoexcitation to the second excited electronic state (S2), excited state population traverses through an S2/S1 CI before being trapped in the excited state minimum of the first excited electronic state (S1). While Losantos et al. did locate a CI between the S1 and ground electronic state (S0), this is energetically inaccessible as it is located above the S1 minimum. This is summarised in Figure 5(a). In contrast, MAA-inspired moieties that are formed of a cyclohexenimine core repopulate S0 following photoexcitation, this time directly to S1: excited state population passes through a now energetically accessible S1/S0 CI, as shown by Figure 5(b). All the systems studied show that the geometry of the CI involves an out of plane movement for both substituents at positions 1 and 3 on the ring (see Figure 4(b)). Furthermore, non-adiabatic molecular dynamics simulations conducted on molecules 6 and 8 (Figure 4(b)) predict an excited state (S1) lifetime of approximately 200 fs. This lifetime is shorter than that of current commercial filters such as menthyl anthranilate and avobenzone.54–56 The shorter excited state lifetime is beneficial to potential sunscreen filters; the probability for competing (reactive) pathways within the sunscreen filter itself, or side reactions between the electronically excited sunscreen filter and additional compounds (within a commercial formulation) is minimised. These competing side reactions can also induce unfavourable reactions some of which have been known to cause skin irritation.11,12
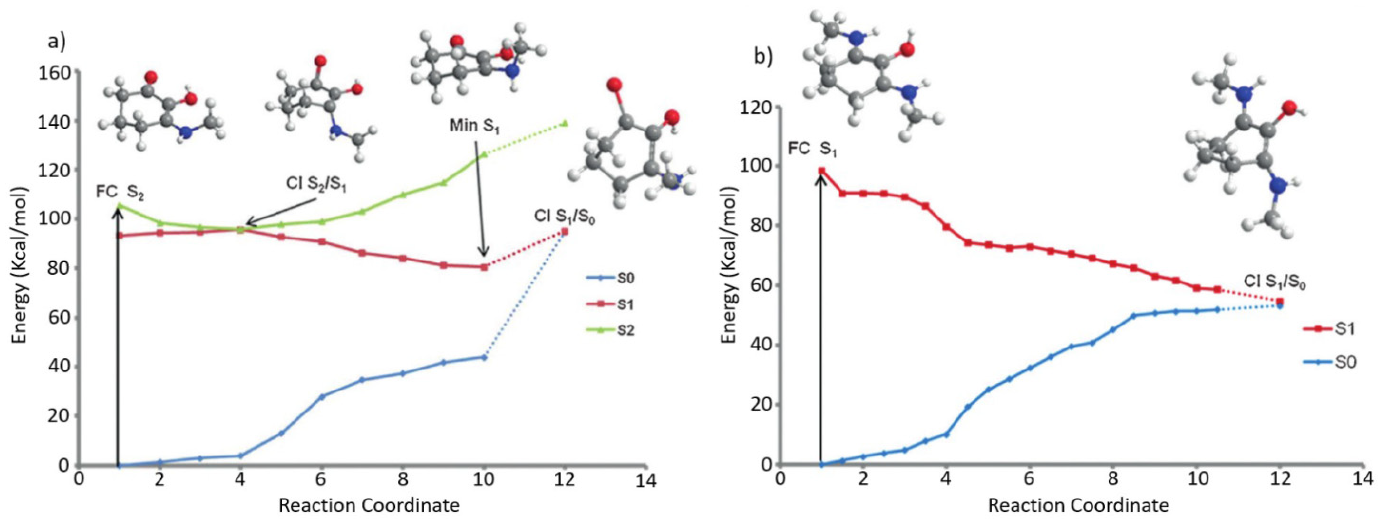
Calculated minimum energy pathways for systems studied by Losantos et al.: (a) structure 1 and (b) structure 6, from Figure 4.
Given their findings regarding the differing core structures, Losantos et al. proceed to suggest 16 additional structures based upon the imine core moiety, each allowing for variation in the electronic ground state absorption to fully cover the UVA and UVB region of the electromagnetic spectrum. They also further explore the photostability and perform solar protection factor (SPF) measurements on mixtures of these systems. 27 These findings are in notable contrast to commercial formulations, demonstrating significant increases in SPF. Of these 16 suggested structures, Losantos et al. have subsequently published a follow-on paper, on three of these proposed structures, employing both transient electronic absorption spectroscopy (see section ‘Transient electronic absorption spectroscopy’) and fluorescence up conversion to monitor the excited state lifetimes. These experimental studies are complemented by CASPT2 calculations (see section ‘Theoretical methods’). 57 Their recent experiments agree with their computational findings (vida supra), and case study 2 of an easily accessible S1/S0 CI promoting rapid repopulation of the electronic ground state. 57
Case study 2
Building on the work by Losantos et al., an experimental investigation into two of the systems, a cyclohexenone and cyclohexenimine core shown as in Figure 6, was performed by Woolley et al. 16 They performed transient electronic absorption spectroscopy (see section ‘Transient electronic absorption spectroscopy’) on both systems in a variety of solvents (acetonitrile and methanol), to unravel the effect of solvent environment on the photoexcited-state dynamics.
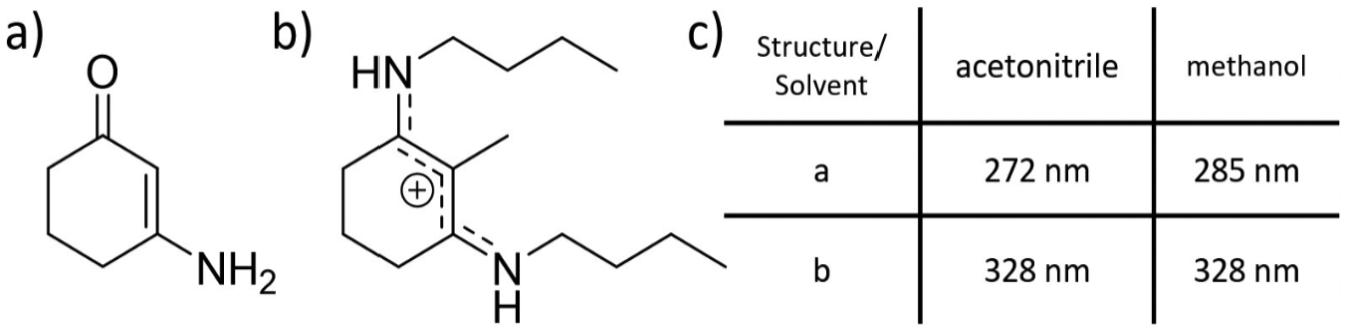
Chemical structures of systems studied by Woolley et al., (a) cyclohexenone core structure and (b) imine core structure. (c) The absorption maximum for each system in the two solvents used in the study.
Woolley et al.’s experimental TAS, shown in the form of false colour heat maps, in Figure 7, revealed that, following photoexcitation at the absorption maximum of the cyclohexenone core (see Figure 6), the vibrationally hot S1 state undergoes vibrational cooling, leading to population of the S1 minimum. Excited state population localised within the S1 minimum is subsequently trapped for at least 2.5 ns (a consequence of the maximum time-window of the experimental setup used by Woolley et al.). On close inspection of the false colour map of Figure 7(a), the initial broad absorption (340–550 nm) is assigned to a initially populated, vibrationally hot, excited state which then vibrationally cools (spectrally blue shifts) on the S1 state. The strong absorption (shown in red), which persists for the maximum time-window of the experiment, describes the trapped excited state population at the S1 minimum. While the change of solvent environment did affect the observed spectra, mainly in terms of signal amplitude (cf. Figure 7(a) and (b)), Woolley et al. concluded that the excited state dynamics followed the same relaxation mechanism regardless of solvent environment, that of excited state population trapped in the S1 state.
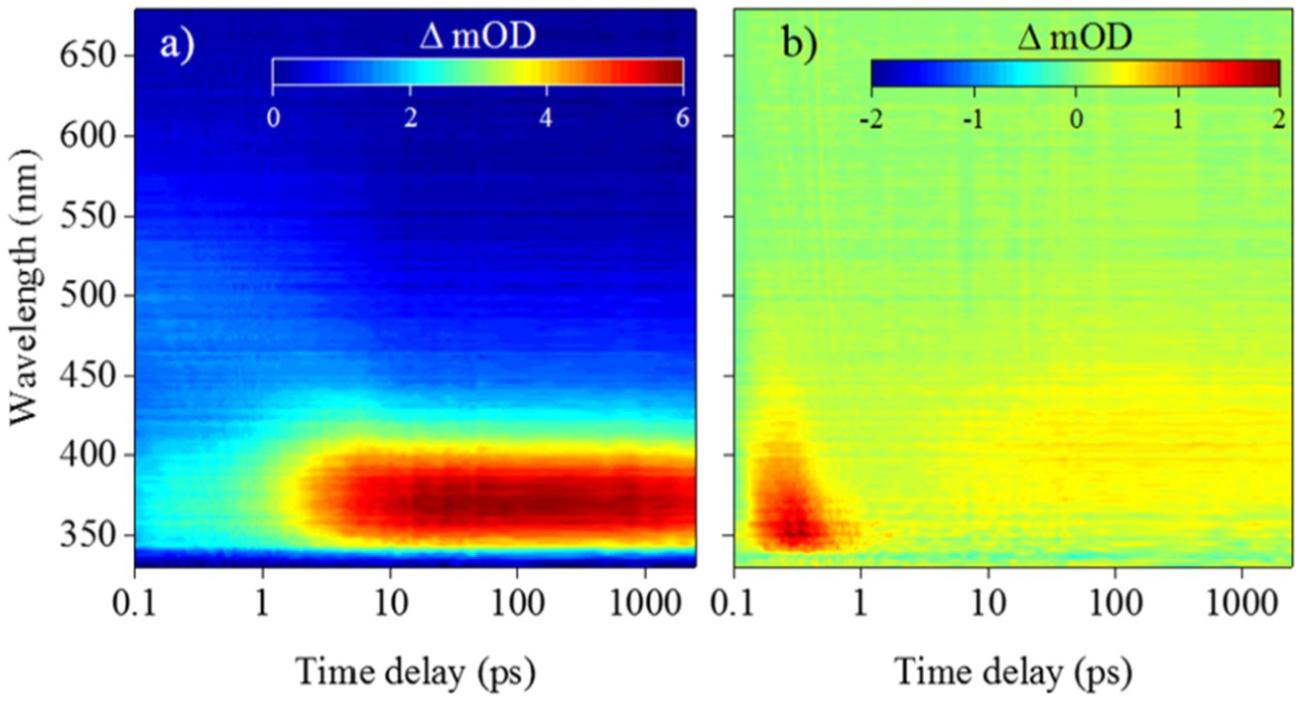
False colour heat maps of transient absorption spectroscopy data collected by Woolley et al. for the cyclohexenone core structure in two solvents: (a) acetonitrile and (b) methanol.
For the cyclohexenimine core photoexcited at the absorption maximum (see Figure 6), the TAS are once again presented as false colour heat maps in Figure 8, display short lived stimulated emission and ground state bleach features (shown in blue) that persist for approximately 500 fs. The data then displays characteristic properties of a vibrationally hot electronic ground state (shown in red): 58 a sharp absorption that rapidly spectrally blue shifts and decays after a few ps. Woolley et al. assign these features to excited state population traversing from the initial Franck–Condon window towards the S1/S0 CI, giving rise to the blue stimulated emission. This is subsequently followed by vibrational energy transfer to the solvent (shown in red), possibly enhanced through the charged nature of the species (see Figure 6(b)).
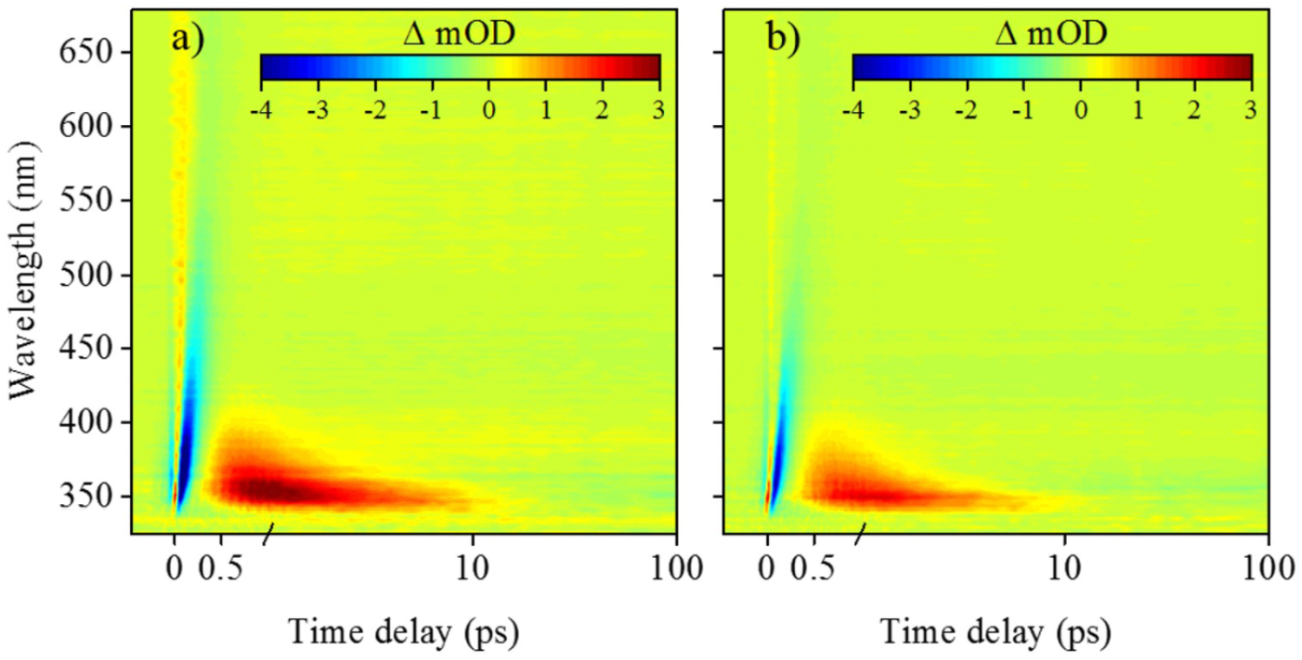
False colour heat maps of transient absorption spectroscopy data collected by Woolley et al. for the imine core structure in two solvents: (a) acetonitrile and (b) methanol.
Overall, the work performed by Woolley et al. corroborates the theoretical and experimental investigations performed by Losantos et al. discussed in case study 1. In particular, the results obtained for the cyclohexenone core unit from both experiment and theory clearly demonstrates that repopulation of the ground electronic state is prevented, shown by the long-lived absorption in the TAS attributed to the S1 state absorption (Figure 7(a)). In the context of a photoprotectant species, the long-lived excited state population is potentially detrimental; the extended lifetime (>2.5 ns) in the S1 state increases the likelihood for competing photochemical processes, for example, triplet state formation (which may subsequently be quenched by triplet oxygen to generate cytotoxic singlet oxygen). Interestingly gadusol and mycosporine-glycine (Figure 1(b)), which have a cyclohexenone core, have been shown to demonstrate high photostability.59–61 This may imply that further modification of the core unit makes the S1/S0 CI energy accessible to excited state population, possibly through additional substitution to differing ring positions (Figure 6(a)). Further work is certainly warranted in this regard, to assess how molecular complexity of the cyclohexenone core influences the photoexcited-state dynamics. Conversely, studies based on the cyclohexenimine core should also be carried out to fully understand the excited state landscape associated with this rapid relaxation.
Case study 3
Case studies 1 and 2, vide supra, focussed on tracking the intramolecular energy transfer within MAA-inspired structures using either theory and computation or time-resolved pump-probe spectroscopy. Work by Conde et al. 62 uses the advantages of photoacoustic calorimetry to follow the transfer of absorbed photon energy through intermolecular interactions between solute and solvent for two common MAAs, shinorine and porphyra-334 shown, respectively, in Figure 9(a) and (b).62,63 Furthermore, Conde et al. conducted steady-state measurements on shinorine to elucidate the photodecomposition and fluorescence quantum yield along with laser flash photolysis to study the triplet state absorption. The values for photodecomposition and fluorescence quantum yield are then compared to the already reported data for porphyra-334. 63
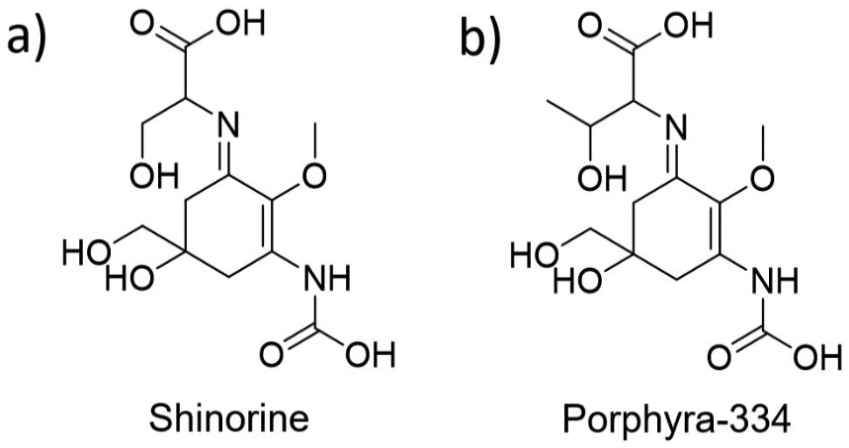
Chemical structures of systems studied by Conde et al.: (a) shinorine and (b) porphyra-334.
Photoacoustic calorimetry performed on both shinorine and porphyra-334 (both excited at 355 nm) showed equivalent values of the initial amplitude, H (see section ‘Photoacoustic calorimetry’ for further explanation), between the solute and the reference sample, indigo carmine. 43 This equivalence (between solute and standard) is an indication that the energy absorbed by the MAAs is rapidly released as heat to the surrounding solvent. Conde et al. 62 note that the percent conversion of this energy transfer (solute to solvent) is 96%–98%, leaving some residual energy trapped within the MAA which undergoes intersystem crossing to populate a triplet state. The authors confirm the presence of the triplet state for both shinorine and porphyra-334 with the use of laser flash photolysis;63,64 this triplet state accounts for the remaining energy trapped within the MAAs.
To corroborate their photoacoustic calorimetry measurements, Conde et al. performed steady-state experiments to fully describe the photodecomposition and relaxation dynamics of shinorine and porphyra-334. These results validate the photoacoustic calorimetry measurements with negligible fluorescence quantum yields determined, with values on the order of 10−4. The rate of photodecomposition was found to be of the same magnitude, leading the authors to attribute a similar relaxation pathway as described for the MAA-inspired structures above (case study 1, Figure 5(b)).
The authors close by placing their research in the wider field of natural sunscreens highlighting that their results strongly favour the use of both shinorine and porphyra-334 in an in vivo environment as natural sunscreen filters, even though a small proportion of the excess energy remains trapped as a triplet population with a significantly longer lifetime.62,63,65 Relating this work to case studies 1 and 2, the proposed overall dynamics of the system agree with those of the MAA-inspired systems by Losantos et al. 16 and Woolley et al. 27 that of rapid relaxation through an accessible CI resulting in a vibrationally excited electronic ground state.
Conclusion and outlook
Given the continuing increase in skin cancer cases and recent concerns raised over current sunscreen filters such as phototoxicity and photdegradation,9,10,13–15,66–68 unravelling photoprotection mechanisms through an intrinsic understanding of the photochemistry of both nature-based (and inspired) and artificial sunscreen filters continues to grow as a field of research.16,27,69 Studies on exploring the synthesis and isolation of MAAs in nature provides complementary aspects of this research.7,70,71 That being said, more work is certainly warranted. Notably, identification of additional MAA-inspired sunscreens is essential: although 30 MAAs have been isolated, regions of the UVR spectrum remain poorly protected from using pure MAA products alone, which is undoubtedly an area where functionalisation of core MAA structures can play a key role.8,27 In addition, a greater photochemical exploration of MAAs should be undertaken to consolidate the relaxation pathways of MAAs, such that we are then best positioned to propose ‘rules’ for efficient molecular design, leading to MAA-inspired sunscreen filters. Undeniably, this will necessitate the use of both experiment and theory.72,73
Finally, it is evident from this review that MAAs present a new and exciting class of photoprotective molecules. However, as illustrated by the above Case Studies, to fully understand their photoprotection capabilities, experimental methods (such as transient electronic absorption spectroscopy and photoacoustic calorimetry) must move towards mimicking a more realistic environment. Sunscreen filters are embedded within a complex formulation which in turn is applied to skin, that is itself a dynamic environment of physiological function. Consequently, the next steps should involve an experimental philosophy that moves away from the traditional analytical methods (e.g. sunscreen dissolved in a solvent) to focus on these new frontiers with the introduction of emollients and selective choice of surface to better model the cosmeceutical applications along with the in vivo nature of the system.
Footnotes
Acknowledgements
J.M.W. is grateful to EPSRC and Newport Spectra-Physics Ltd for a joint studentship. V.G.S. thanks the HO2020 FET-OPEN Grant BoostCrop for financial support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.