
Abstract
PulseNet, the National Molecular Subtyping Network for Foodborne Disease Surveillance, was established in 1996 through a collaboration with the Centers for Disease Control and Prevention; the US Department of Agriculture, Food Safety and Inspection Service; the US Food and Drug Administration; 4 state public health laboratories; and the Association of Public Health Laboratories. The network has since expanded to include 83 state, local, and food regulatory public health laboratories. In 2016, PulseNet was estimated to be helping prevent an estimated 270 000 foodborne illnesses annually. PulseNet is undergoing a transformation toward whole-genome sequencing (WGS), which provides better discriminatory power and precision than pulsed-field gel electrophoresis (PFGE). WGS improves the detection of outbreak clusters and could replace many traditional reference identification and characterization methods. This article highlights the contributions made by public health laboratories in transforming PulseNet’s surveillance and describes how the transformation is changing local and national surveillance practices. Our data show that WGS is better at identifying clusters than PFGE, especially for clonal organisms such as Salmonella Enteritidis. The need to develop prioritization schemes for cluster follow-up and additional resources for both public health laboratory and epidemiology departments will be critical as PulseNet implements WGS for foodborne disease surveillance in the United States.
The United States has one of the safest food supplies in the developed world, yet the burden of foodborne disease is high. The Centers for Disease Control and Prevention (CDC) estimates that more than 9 million episodes of foodborne illness resulting in more than 55 000 hospitalizations and more than 1300 deaths occur annually in the United States. 1 For more than 20 years, PulseNet, the National Molecular Subtyping Network for Foodborne Disease Surveillance, has been on the forefront of detecting local clusters and multistate foodborne outbreaks using pulsed-field gel electrophoresis (PFGE) to characterize isolates from patients, food, and the environment (Figure 1). 2 The network of public health laboratories within PulseNet uses standardized laboratory and data analysis methods in addition to a communication platform to quickly detect clusters of cases and link potential food and/or environmental sources. The impact of PulseNet is most evident in the timeliness of outbreak detection and the number of cases per outbreak identified. For example, in 1993, before PulseNet, it took public health officials 39 days to detect an outbreak of Escherichia coli O157 in western states, resulting in more than 720 illnesses and 4 deaths. In 2002, it took only 18 days for PulseNet to detect an E coli O157 outbreak in Colorado, resulting in 44 illnesses and no deaths. 3 In 2016, an economic evaluation of PulseNet showed that the network prevents an estimated 270 000 illnesses caused by Salmonella, Listeria monocytogenes, and E coli O157 annually in the United States, resulting in an estimated $507 million saved in medical costs and lost productivity annually. 4
Map of PulseNet USA participating laboratories and their corresponding 7 regions. PulseNet is a national laboratory-based surveillance network that uses DNA fingerprinting method, including whole-genome sequencing, for outbreak cluster detection and surveillance of foodborne bacterial pathogens. Used with permission from the Centers for Disease Control and Prevention. 2
Emerging technologies, specifically next-generation sequencing (NGS) methods, are changing clinical diagnostics and public health laboratory practice. The costs associated with sequencing whole genomes have decreased dramatically in recent years, with manufacturers producing relatively low-cost, high-throughput instruments. When the first NGS platform was introduced in 2005, the cost to sequence a 5-megabase pair-sized bacterial genome was about $5000. Today, it is about $120. 5 This cost is higher than that of PFGE ($30), but as mentioned later, WGS will also replace many traditional reference characterization methods, making it cost-efficient to use in public health laboratories. The terms NGS and WGS are often used interchangeably even though, strictly speaking, NGS refers to the sequencing technology, whereas WGS refers to the entire sequencing and analysis process. However, the greatest advantage of NGS technology to the public health laboratory system is that it offers a universal subtyping and reference characterization system. That is, the same technology can be applied to bacteria, viruses, and other pathogens; therefore, laboratories can efficiently use NGS for many public health surveillance programs. In the long term, NGS will likely provide cost savings because many of the traditional reference-based public health methods, such as pathogen identification, virulence, and antimicrobial resistance testing, can be consolidated into a single genomic workflow.
In addition, changes in clinical diagnostics, specifically the development of culture-independent diagnostic tests, have affected public health laboratory practice. 6 -8 Several of these culture-independent diagnostic test platforms are available to quickly identify gastrointestinal, respiratory, and bloodstream infections. These new tests allow clinical laboratories to detect agents directly from primary specimens within hours. No culturing of the organism is required to identify the pathogen, which offers the advantage of performing these tests on site in emergency, urgent-care, and other point-of-care settings. The disadvantage to public health of the widespread use of culture-independent diagnostic tests is that isolates are no longer recovered and forwarded to public health laboratories for national isolate-based surveillance programs such as PulseNet. Because of these emerging trends in clinical diagnostics and biotechnology fields, PulseNet is transitioning surveillance toward sequencing DNA and eventually sequencing DNA from pathogens directly in primary specimens (metagenomics). This transition will allow PulseNet to continue to adapt to both advances in biotechnology and changes in clinical laboratory practices and diagnostics.
PulseNet’s Transition to WGS for Surveillance
Pulsed-field gel electrophoresis has been PulseNet’s primary subtyping method for detecting clusters of Salmonella, Listeria monocytogenes, Shiga-toxin producing E coli (STEC), Vibrio, Shigella, and Campylobacter subspecies from clinical, food, and environmental sources. The DNA fragment-banding pattern generated through PFGE provides a DNA fingerprint that enables public health scientists to compare different isolates. Databases, developed by and housed at CDC, allow public health laboratories across the country to submit DNA fingerprint data and associated supporting metadata to CDC in real time for rapid cluster detection and surveillance. Some limitations of PFGE subtyping include the need for standardized protocols for each pathogen of interest, limited discriminatory power compared with WGS, and the inability to determine true phylogenetic relatedness between and among isolates.
Notably, PulseNet’s decision to implement WGS for surveillance also included the sharing of pathogen-read sequences publicly in real time and in the GenomeTrakr database of foodborne pathogens, via the National Center for Biotechnology Information (a branch of the National Institutes of Health that provides access to various biomedical and genomic information). These sequences are available to academicians and public health officials for further analysis. Two analytical approaches are commonly used for pathogen subtyping using WGS data: single nucleotide polymorphism (SNP) analysis and multi-locus sequence typing (MLST). SNP analysis compares base changes at any position in the genome of a test strain with a closely related reference strain sequence (Figure 2A). MLST assesses any differences in genes (loci) of test strains compared with a genus-specific reference database of sequence variants (alleles) (Figure 2B). When multiple isolates are compared by these methods, the number of SNP differences or allelic differences is counted in a pairwise fashion and a similarity matrix is generated. For SNP data, a phylogenetic tree based on nucleotide differences can also be generated.

Examples of 2 analytical approaches commonly used with whole-genome sequencing (WGS) data. A) The single nucleotide polymorphism (SNP) approach assesses and compares base changes at any position in the genome of a test strain with a closely related reference strain sequence. A read represents a fragment of sequence data being compared in the analysis. All bases are different when compared with the reference genome, with 9 of 10 reads representing a single base change to a “T” (highlighted in bold). Read 5 represents a potential sequencing error at the underlined position. This highlights the need for cutoffs for base frequency when calling an SNP. B) Multi-locus sequence typing assesses and compares differences (highlighted in bold) in genes (loci) of test strains with a genus-specific reference database of sequence variants (alleles).
To visualize differences, a hierarchical tree may be drawn based on this similarity matrix. Isolates that show few SNP or allelic differences are more related and have a more recent common ancestor than isolates that have a greater number of SNPs or allelic differences. For both SNP and MLST analyses, either the whole genome or the core genome (part of a genome shared among compared test genomes) can be assessed, yielding differing levels of resolution and marker stability. Isolates from single-source outbreaks typically have between 0 and 15 SNP or allele differences. This range of differences depends on the clonality of the organism in question and the epidemiologic context. Clonal organisms have stable genomes that show little to no variation over time. Non-clonal organisms have highly variable genomes that can mutate in a short period of time, even in a matter of weeks. Hence, if a long-lasting outbreak caused by a non-clonal organism occurs, the variation among outbreak isolates can be well above the 15 SNP or allele differences. Both MLST and SNP analyses require a reference; in MLST, the reference is a database of allelic variants at all loci of a given species, whereas the reference for SNP analysis is a single genome of a strain that is closely related to the isolates to be subtyped, such as outbreak isolates. MLST can be standardized by all users using the same allele database and the results shared in a network of laboratories.
For this reason, as of summer 2019, whole-genome MLST is the primary subtyping approach in PulseNet, and work is ongoing to expand this standardization globally with allele databases and analytic tools shared in the public domain. 9 Because the 2 analytic approaches differ, SNP analysis and MLST are complementary methods; in PulseNet, SNP analysis is sometimes used to confirm results generated by MLST. Another reason for using MLST in PulseNet is that with MLST, sequences may be named in a phylogenetically relevant and hierarchical way with just 1 nomenclatural scheme, which also can be standardized internationally with each species. Such strain nomenclature systems were originally developed for SNP analysis. 10 However, to cover an entire species, numerous reference strains have to be used to correctly identify the SNPs, making it challenging to standardize sequence nomenclature among laboratories. MLST-based strain nomenclature (allele codes) ensures unambiguous communication during outbreak investigations (eg, which isolates are part of the outbreak) and simplifies queries of subtypes in surveillance databases.
A major advantage of WGS compared with PFGE is the ability to determine the genus, species, serotype, virulence profile, antimicrobial resistance, plasmid profile, and other genetic information from the sequence of the microorganism’s genome. WGS makes it possible to replace numerous strain characterization methods with a single cost-efficient WGS workflow that generates more information about the isolates than traditional methods. For example, public health scientists can now use genotyping tools and reference databases to predict the antimicrobial resistance profile for all strains during an outbreak investigation. In the past, because of the labor-intensive nature and high cost of phenotypic antimicrobial resistance testing, it was performed only for a few representative isolates from a given investigation. The real-time antimicrobial resistance information is important for outbreak investigations and, moving forward, will likely have implications for making decisions about patient management and will help investigators increase their understanding of the emergence and spread of resistance genes in pathogens throughout the food safety system. Similarly, information about virulence genes will be extracted for all strains, and this information can be used to prioritize outbreak investigations by focusing on outbreaks caused by the most virulent pathogens and can help researchers understand microbiologic risk factors for various clinical presentations.
Use of WGS in Surveillance for Salmonella Enteritidis in New York State and Minnesota
In 2013, the New York State Department of Health and Minnesota Department of Health began using WGS for surveillance and cluster detection of Salmonella enterica serovar Enteritidis (SE). SE was selected for early implementation of WGS-based surveillance because it is the most common Salmonella serovar in both states (about 30% of all Salmonella) and its genetic uniformity leads to poor cluster resolution by PFGE. In both states, the top 5 PFGE patterns of SE isolates harbor endemic patterns that are too common in the population to be useful in directing epidemiologic investigations.
Two pilot studies demonstrated that these common PFGE patterns seen in SE could be subdivided by WGS using reference-based high-quality single nucleotide polymorphism (hqSNP) analysis. 11,12 From October 2013 through October 2015, New York State evaluated all clinical SE samples received at the laboratory by using an in-house pipeline developed at the New York State Department of Health Wadsworth Center; this analysis resulted in the 5 most common (endemic) PFGE patterns being subdivided into 108 genomic clusters. To limit the number of clusters to be investigated by epidemiologists to those with the highest likelihood of being solved, the following thresholds were implemented: 3 isolates collected within 60 days that differed by no more than 5 SNPs. Based on these filters, 20 genomic clusters were identified in 2017. The large number of potentially informative clusters detected by WGS compared with PFGE further emphasizes the need to consider epidemiologic information before investing resources in a cluster investigation.
Concurrent SE Outbreaks in New York State and Virginia Correctional Facilities
During spring 2016, the Virginia Division of Consolidated Laboratory Services reported to PulseNet that it was investigating a cluster of 3 SE isolates with PFGE pattern JEGX01.0021 associated with the same correctional facility. Simultaneously, the New York State Department of Health was investigating another SE cluster, also with PFGE pattern JEGX01.0021, occurring in a county jail in New York State. As part of a multistate collaboration, New York State and Virginia compared the outbreak clusters from both states by using WGS results generated by the New York State Department of Health Wadsworth Center’s in-house hqSNP pipeline. This hqSNP analysis indicated that the outbreak strains were closely related within each state (0 and 0-2 SNP differences for Virginia and New York State, respectively), but the 2 state clusters were 21 to 22 SNPs from each other, indicating that they were unlikely to be from a common source (Figure 3). As a result, no additional resources were used to find a common source of the 2 outbreaks.

Phylogenetic tree from a case study of concurrent Salmonella Enteritidis outbreaks in correctional facilities in New York State and Virginia. The high-quality single nucleotide polymorphism analysis shows that outbreak strains were closely related within each state, but the 2 clusters were unlikely to be from a common source.
Using WGS to Identify a Salmonella Enteritidis Outbreak with a Common PFGE Pattern
From August through September 2014, 19 cases of SE in Minnesota were identified with PFGE pattern JEGX01.0004/JEGA26.0002 (XbaI/BlnI), a common PFGE pattern in Minnesota that comprises approximately 45% of the SE isolates in the state. WGS was performed simultaneously with PFGE. WGS identified a cluster of 8 isolates that were 0 SNPs different from each other. Local epidemiologists investigated the cluster, and all persons with SE were interviewed. Of the 8 persons who shared the same WGS profile, 6 confirmed consumption of a frozen, stuffed chicken product, 1 indicated possibly consuming the product (frozen chicken product included in food history but brand name unknown), and 1 was a secondary case (likely infected by another person in the outbreak). None of the 11 persons who did not share the same WGS profile had consumed the frozen, stuffed chicken product. The public health laboratory cultured the frozen, stuffed chicken product, and SE with the same WGS profile was isolated from the product (Figure 4). This investigation led to a product recall that likely prevented additional cases of human illness. WGS provided additional discrimination compared with PFGE. Most important, the cluster identified by using WGS allowed epidemiologists to identify a common source.
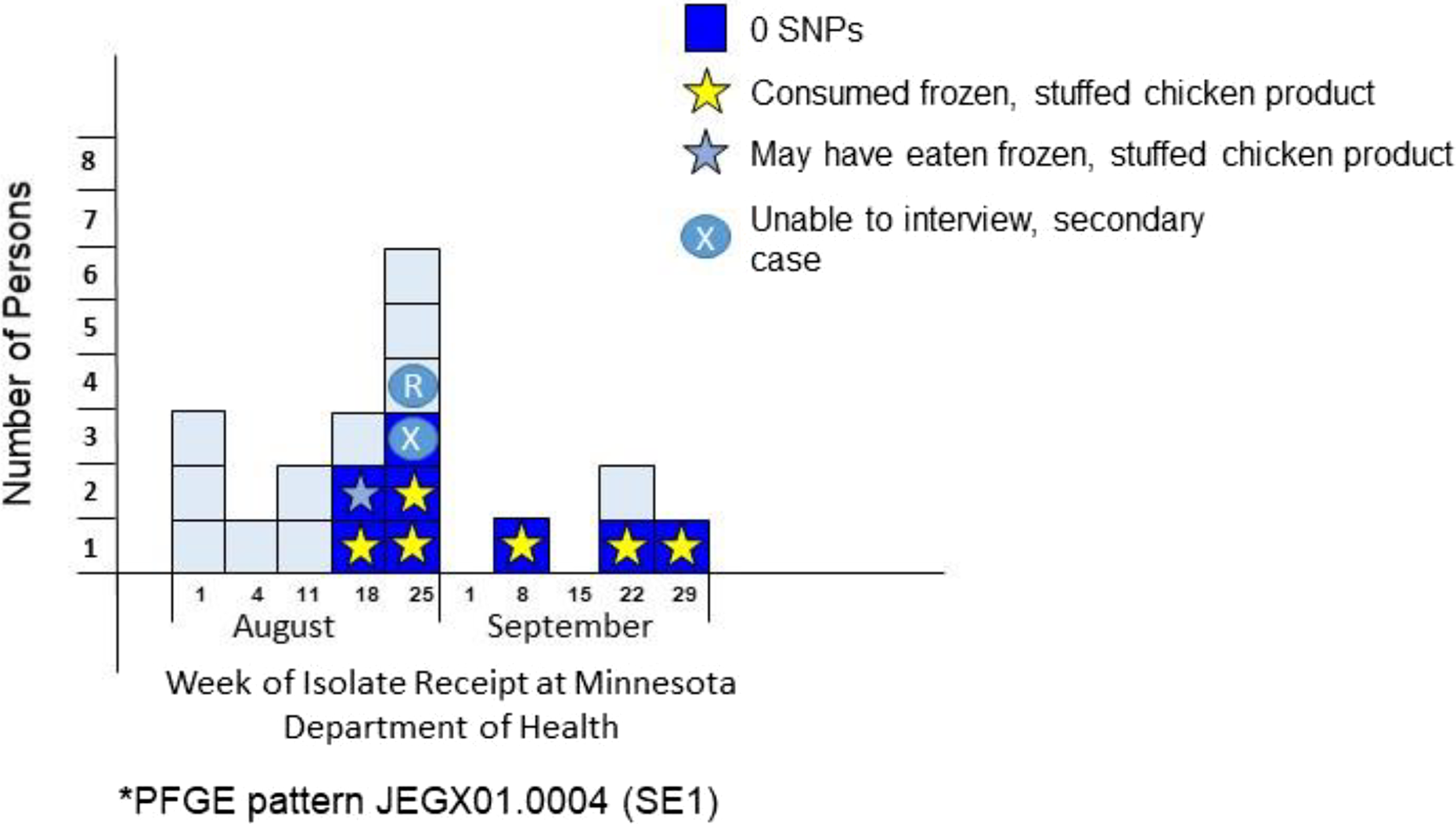
Case distribution of Salmonella Enteritidis pulsed-field gel electrophoresis (PFGE) and single nucleotide polymorphism (SNP) profiles related to frozen, stuffed chicken products in Minnesota, August–September 2014. Each box represents 1 isolate of Salmonella Enteritidis Minnesota PFGE pattern designation SE1B1 received at the Minnesota Department of Health in August and September 2014 (n = 19). High-quality SNP (hqSNP) analysis was performed on all isolates. For this investigation, whole-genome sequencing data (hqSNP profiles) provided additional discriminatory power compared with PFGE alone, which proved useful for this investigation.
Federal Perspectives
Since 2014, the US Food and Drug Administration (FDA) has enhanced its sampling of the portion of the food supply it regulates. In 2013, the FDA initiated the GenomeTrakr network, which includes all FDA field laboratories, selected state agricultural and public health laboratories, and some academic and private laboratories. 13 GenomeTrakr created a database of genomes of pathogens from food produced in the United States and abroad. The database is housed at the National Center for Biotechnology Information and uses tools available in its Pathogen Detection Portal as a first pass to detect isolates of particular interest to food regulators (eg, by matching isolates from the food supply to clinical isolates). 14 The FDA downloads isolate sequences of interest and confirms their relationship by using the FDA SNP pipeline. Further action depends on the situation but often includes the FDA working with public health partners at CDC and in the 50 states to define the scope of the outbreak and to trace the food product to its source for possible regulatory action. WGS has particularly enhanced the detection and investigation of retrospective outbreaks, in which isolates are first recovered from the food supply and sequenced; their sequences then lead to the detection of matching current or historical clinical isolates. These outbreaks are often small when detected but are nevertheless important to investigate because they point to neglected or previously unrecognized risks in food production.
An example of such a retrospective outbreak is the Listeria monocytogenes outbreak linked to consumption of stone fruits (ie, fruit with pits, such as peaches and plums) in 2014. 15 The outbreak was initially detected by PFGE by matching numerous Listeria isolates from stone fruits to 4 patient isolates. Further analysis by WGS indicated that only 2 patient isolates were related to the stone fruit isolates. Interviews determined that those 2 patients had consumed stone fruits, whereas the 2 patients who were excluded (based on WGS) had not consumed stone fruits. Three lessons could be drawn from this investigation: (1) stone fruits could be a source of listeriosis, (2) WGS was better than PFGE at pinpointing the outbreak-related patient isolates, and (3) epidemiologic follow-up of cases is a necessary component to implicate a specific food in an outbreak.
The substantial overlap in the membership of the GenomeTrakr and PulseNet networks is one of the strengths of both, enhancing cluster detection and outbreak investigations. Aside from supporting PulseNet-related surveillance activities with WGS of enteric pathogens from food and environmental sources, GenomeTrakr serves as a resource for preventive controls and traceability by monitoring the persistence and emergence of pathogens in food production and processing environments and potentially linking pathogens to a particular food or facility source. PulseNet detects outbreaks from food and non-food sources through the detection of clusters of closely genetically related clinical isolates and works closely with epidemiologists, food regulators, environmental scientists, and other stakeholders throughout outbreak investigations. Coordination among the networks is accomplished through efforts of the Interagency Collaboration on Genomics for Food and Feed Safety (Gen-FS) and the Global Microbial Identifier Network. The Gen-FS is a US government collaboration of CDC, the US Department of Agriculture, the FDA, and the National Center for Biotechnology Information and involves a steering committee and workgroups for harmonizing various activities among the networks, such as laboratory and analysis methods, sequence quality assessment, proficiency testing, training, and communications. 16 The Global Microbial Identifier Network aims to harmonize many of the same activities on a global scale. It includes about 260 experts from 50 countries representing government agencies, academia, and industry. 17
Conclusions and Future Challenges
As of July 15, 2019, PulseNet transitioned to WGS as the new gold standard method for foodborne diseases surveillance within the network in the United States. As of September 25, 2019, 63 laboratories in 48 states were certified to submit sequence data to PulseNet, and all 50 states had sequencers. Certification in WGS is a quality assurance requirement for laboratories, allowing them to upload data and access national WGS databases for the detection of clusters. On January 15, 2018, PulseNet officially transitioned Listeria monocytogenes real-time surveillance from PFGE to WGS. One of the main barriers to full WGS implementation for the network has been building capacity at local and federal levels. Procurement of sequencers, training public health laboratory workforce, and building an information technology infrastructure for the analytical tools have also been challenging.
Examples from state public health laboratories demonstrate the power and utility of WGS technology to detect foodborne outbreaks, especially in cases of highly clonal serotypes such as Salmonella Enteritidis. As PulseNet transitions to WGS for the remaining foodborne pathogens, Vibrio species, Yersinia enterocolitica, and Campylobacter, the number of clusters detected by local and state public health laboratories will likely dramatically increase and the number of cases per cluster will likely decrease. 18 Strong epidemiologic information and strain characteristics from WGS (ie, resistance or virulence factors) will be increasingly essential for solving clusters of foodborne diseases being investigated by PulseNet laboratories.
Although WGS offers the highest resolution among current DNA fingerprinting technologies, will it provide adequate resolution for highly clonal organisms tested on a national scale, such as Salmonella Enteritidis? At the time of this writing, examination of hqSNP trees for Salmonella Typhimurium and Salmonella Enteritidis at the National Center for Biotechnology Information revealed 6 trees that harbored more than 2000 samples that were within 50 SNPs of each other. In cases in which clonality is confounding the identification of the specific microorganism’s strain responsible for an outbreak, even for WGS data, it will be imperative to be able to overlay epidemiologic and additional WGS information (ie, plasmid, phage, virulence, or resistance data) that is often excluded from WGS phylogenetic analysis to focus an investigation adequately.
The increasing use of culture-independent diagnostic tests by clinical laboratories may become a challenge for the future use of WGS for surveillance in PulseNet. These methods provide public health investigators with more reliable data about pathogens rarely diagnosed with traditional culture-based methods, such as diarrheagenic E coli (apart from STEC), Yersinia, and Vibrio. 19 However, adoption of these tests in clinical laboratories results in either a reduction in the number of isolates received by public health laboratories or an increase in primary samples submitted to public health laboratories for culture isolation of the pathogen. Current public health testing methods, including WGS, require isolates to detect and investigate outbreaks and monitor trends in antimicrobial resistance and pathogen incidence. This information is necessary to help prioritize and document the effect of mitigation efforts to prevent foodborne infections. PulseNet is in a race against time to develop diagnostic culture-independent methods directly from primary samples (metagenomics approaches) that will provide public health officials with subtyping and other information necessary for public health action.
Footnotes
Acknowledgments
The authors thank the Wadsworth Center Applied Genomic Technologies Core and the Wadsworth Center Bioinformatics Core for sequencing and analyzing, respectively, isolates reported in Figures 3 and ![]() .
.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This publication was supported by cooperative agreement no. 5NU60OE000103 funded by the Centers for Disease Control and Prevention (CDC). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of CDC or the US Department of Health and Human Services. This project was 100% funded with federal funds from a federal program of $2.2 million.