
Abstract
The in situ polymerase chain reaction (PCR) is a technique that has important applications in the diagnosis of viral and bacterial diseases. This study investigated an in situ PCR assay established to detect the presence of Chlamydia trachomatis in endocervical swabs. In addition, histological sections of endocervical squamous cell carcinoma were analyzed because previous studies had revealed a significant association with C. trachomatis. A total of 20 cervical neoplasms (squamous cell carcinoma in situ; n = 10; invasive squamous cell carcinoma; n = 10) and endocervical smears taken from five patients with and without inflammatory changes were analyzed by conventional PCR. Chlamydial DNA was found in 10 histological samples (six carcinomas in situ, four invasive carcinomas) and in one endocervical swab from a patient with known C. trachomatis infection. Positive specimens were used for establishing an in situ PCR assay (IS-PCR). After IS-PCR, these samples showed dense cytoplasmic staining of endocervical cells (smears) and non-neoplastic epithelial cells (cervical neoplasms). The other tumor samples and smears did not demonstrate positive PCR reaction. The results indicate that in situ PCR is an effective technique for localizing C. trachomatis in target cells because IS-PCR detection of chlamydial DNA correlated with histological and cytological features.
Keywords
Chlamydia trachomatis is a pleomorphic nonmotile organism about 0.2–1.5μm in length. This obligate intracellular bacterium depends on host cell metabolites and shows a unique growth cycle characterized by formation of reticulate bodies, intermediate bodies, and elementary bodies (Hodinka et al. 1988). C. trachomatis occurs in at least 18 serovars that can be differentiated by the nucleotide sequence of the major outer membrane protein, MOMP (Lampe et al. 1993). C. trachomatis serovar A-C is the causative agent of trachoma, a hyperendemic disease that occurs in less developed countries and leads to conjunctivitis and blindness (Mardh et al. 1982). C. trachomatis serovar L1-L3 causes lymphogranuloma venereum, a rare venereal disease (Caldwell and Kuo 1977). Infections with C. trachomatis serovar D-K are the most common sexually transmitted diseases worldwide (Hoyme 1989). Affected patients develop signs of urethritis associated with epididymitis and prostatitis (Oriel 1992). The disease is often asymptomatic and is characterized by local infections that tend to ascend from the cervix or urethra into the endometrium and the Fallopian tubes of infected women (Rice and Schachter 1991). Without antibiotic treatment, long-term infections may result in chronic manifestations complicated by infertility, obliteration of the Fallopian tubes, or ectopic pregnancy (Brunham et al. 1985).
For a long time, the cell culture method established 30 years ago was the technique of choice to detect the intracellular bacteria (Ripa 1982). This method was succeeded by techniques such as the direct immunofluorescence test (DIF, Syva Micro Trak) and enzyme immunoassays (EIA), which can be routinely applied to large pools of samples (Domeika et al. 1994). However, some of these antigen tests yield low detection rates (Eley et al. 1992). The polymerase chain reaction (PCR) is a more sensitive technique and has proved that low copy numbers of the serovars D and I are frequently found in asymptomatic women (Lan et al. 1995). The method was also successfully used in combination with restriction fragment length polymorphism to amplify the bacterial MOMP-1 gene to differentiate the serovars of C. trachomatis (Frost et al. 1991).
In situ PCR is a novel method that has been already used to detect low copy number DNA sequences of viral HIV, HPV, CMV, and HBV in tissue (Komminoth and Long 1993). In this study the technique was used to detect C. trachomatis in cells of gynecological smears and in histological samples obtained from female patients with intraepithelial neoplasia or invasive squamous cell carcinoma. Both tumors were formerly reported to be associated with C. trachomatis infection (Paavonen et al. 1979; DeSanjose et al. 1994; Nunez-Troconis 1995). To our knowledge, this is the first report on the use of IS-PCR to detect C. trachomatis in human cells.
Materials and Methods
Sample Collection
Paraffin-embedded squamous cell carcinomas of the cervix were collected from 20 patients aged 21–36 years. The tumors were histologically classified as squamous cell carcinoma in situ (n = 10) and invasive squamous cell carcinoma (n = 10) and were obtained from 1995 to 1996 by coldknife conization. In addition, cervical smears from five patients with or without inflammatory cellular changes were analyzed. Two of the five smears were obtained from patients with C. trachomatis infection. The remaining swabs were from patients who were not infected. The endocervical samples were obtained by inserting a cotton swab, rotating it 360° for 10 sec, and transferring the cells to a glass slide. The sample was fixed with 96% ethanol (v/v) before transport. Positive controls were McCoy mouse fibroblasts infected with C. trachomatis L2. A melanoma cell line served as the negative control.
PCR
DNA preparation. Histological sections and cervical smears were scraped into sterile Eppendorf microfuge tubes using a clean razor blade. The histological samples were incubated twice at 37C for 5 min with xylene to remove the residual paraffin. The resulting pellets were used for DNA isolation by applying the QIAamp Tissue Kit (QIAGEN; Hilden, Germany). The eluted DNA was concentrated by vacuum centrifugation, suspended in 3 μl H2O, and stored at −20C.
Amplification. For amplification of the C. trachomatis plasmid sequence, the PCR primers were 5′-GGACAAATCGTATCTCGG-3′ (T1, sense primer) and 5′-GAAACCAACTCTACGCTG-3′(T2, antisense primer) (Mahony et al. 1992). The combination yielded a 517-
The standard PCR mix consisted of 2.75 mM MgCl2 (Perkin-Elmer; Weiterstadt, Germany), 10 × PCR buffer [Perkin-Elmer; contains 500 mM KCl, 100 mM Tris-HCl, pH 8.3, at 25C, 0.3 μg primer, 10 μM of each dNTP (Pharmacia; Freiburg, Germany), 1 U of Taq DNA polymerase (Perkin-Elmer)]. One μl of extracted DNA was used for PCR. The final volume was 50 μl. The mixture was overlaid with mineral oil (Sigma; Munich, Germany). Thermal cycling was performed on a DNA thermal cycler 480 (Perkin-Elmer) using thin-walled reaction tubes. Standard precautions against cross-contamination were followed (Kwok and Higuchi 1989). Amplified DNA was separated for 2 hr at 80 V on a 3% (w/ v) agarose gel (Biozym; Hessisch Oldendorf, Germany) containing 0.5 μg ethidium bromide/ml.
In Situ PCR
Fixation and Digestion of Material. The paraffin-embedded cervix carcinoma specimens were cut in 4-μm sections, mounted on 3-aminopropyl-triethoxysilane-coated glass slides (Perkin-Elmer), and incubated overnight at 60C in an incubator. The sections were dewaxed by washing twice in xylol at 37C for 5 min. Residual xylol was extracted by placing the samples in 80, 90, and 100% ethanol for 1 min. Tissue was air-dried and rehydrated by placement in 100, 90, and 80% ethanol for 1 min. The slides were washed with 1 × PBS buffer (150 mM NaCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.1) for 5 min and transferred into TEN/protease A buffer (1 M Tris-HCl, pH 8.0, 0.5 ml/0.5 M EDTA, pH 8.0 0.1 ml/5 M NaCl 1.0 ml/ 6μg protease/ml final concentration) for 10 min. The specimens were washed with 1 × PBS for 5 min and air-dried.
The cervical smears placed on coated glass slides were fixed in 100% ethanol and air-dried. The swabs were fixed overnight in 1 × PBS/formaldehyde 4% (v/v), washed with 1 × PBS for 5 min, and air-dried. TEN/protease A buffer (1 M Tris-HCl, pH 8.0, 0.5 ml/0.5 M EDTA, pH 8.0, 0.1 ml/5 M NaCl 1.0 ml/800 ng protease/ml final concentration) was pipetted onto the cells. The cells were incubated at room temperature (RT) for 5 min, washed with 1 × PBS, and airdried.
Amplification. PCR amplification was conducted on an in situ thermal cycler (Perkin-Elmer). The PCR mix was trapped over the air-dried samples by silicon diaphragms using mounting clips and an assembly tool (Perkin-Elmer).
The selection of cycling conditions depended on the type of material. For amplification of DNA isolated from histological sections, reaction tubes were heated to 94C for 7 min followed by 40 cycles at 95C for 1 min, 60C for 1 min, and 72C for 1.5 min and a final extension at 72C for 7 min. For amplification of DNA isolated from cytological smears, reaction tubes were heated to 94C for 7 min followed by 40 cycles at 95C for 1 min, 55C for 45 sec, and 72C for 1 min and a final extension at 72C for 7 min.
The PCR mix consisted of 2.75 mM MgCl2 (Perkin-Elmer) 10 × PCR buffer [Perkin-Elmer; contains 500 mM KCl, 100 mM Tris-HCl, pH 8.3, at 25C, 0.3 μg primer, 10 μM of each dNTP (Pharmacia), 0.5 nM biotin-16-dUTP (Boehringer; Mannheim, Germany), 5 U Taq DNA (Perkin-Elmer)]. Polymerase PCR was performed on a final volume of 50 μl. The mix contained 0.3 μg of the primers T1 and T2 for a 517-
Detection of Biotin-labeled PCR Fragments
Labeled DNA fragments were detected with primary antibody, anti-biotin monoclonal antibody immunoglobulin (Boehringer) at 1:200 dilution and using the Universal immunostaining kit streptavidin-peroxidase with ACE (Immunotech; Coulter, Marseille, France) according to the manufacturer's instructions, with the following modification. Before adding the primary antibody, sections and smears were incubated with drug-free serum (Bio-Rad; Munich, Germany) at RT for 2 hr. The recommended incubation time with primary antibody was reduced from 1 hr to 30 min.
Controls
Appropriate controls were performed as recommended by Komminoth and co-workers (1992). These included the use of (a) irrelevant primers for amplification (primers directed against the outer surface protein OspA of the bacterium Borrelia burgdorferi; Barbour and Garon 1988), (b) positive and negative control samples, (c) solution-phase PCR on the extracted DNA of the cervical swabs or cervical sections, (d) omission of primary antibody in the immunochemical detection of biotin, (e) IS-PCR with positive McCoy cells or cervical swabs in suspension, (f) omission of the primers T1/T2, (g) omission of Taq DNA polymerase, (h) negative control antibody in place of the primary antibody (see data sheet of staining kit), (i) κ/λ positive controls (an antibody solution provided in the kit). The test specimens were used as an internal control. The antibody reacted with plasma cells in the sections. Alternatively, dewaxed sections of lymph nodes from patients with centroblastic lymphoma (Kiel classification) served as positive controls that were stained with the solution. In each run, diffuse cytoplasmic staining of target cells proved that the test was done correctly and that the fixation was at least sufficient and appropriate.
Sequence Analysis of C. trachomatis DNA Fragment Obtained by IS-PCR
Several histological sections (15μm each) were cut from a cervical sample that had shown C. trachomatis positivity according to the results of conventional and IS-PCR. The sections were used for IS-PCR. After amplification, the sections were pooled and washed with 1 × PBS for 5 min. The DNA was isolated as described above, concentrated in a vacuum concentrator (Eppendorf; Hamburg, Germany), and separated on 3% low melting point agarose (Biozym). A cube of agarose containing a 517-
Electron Microscopy
McCoy cells infected with C. trachomatis and cells from fresh cervical smears were harvested by centrifugation at 1000 × g for 10 min and fixed in a solution of glutaraldehyde 0.3% (v/v) (grade I; Sigma) in PBS at 4C for 1.5 hr. The cells were embedded in the low-temperature resin Lowicryl K4M (Chem Werke Lowi; Waldkraiburg, Germany) according to Carlemalm et al. (1982). Ultrathin sections were cut from the samples and poststained with uranyl acetate (pH 4.8). Electron micrographs were taken with a Philips EM 301 (Philips; Eindhoven, Netherlands).
Results
Detection of C. trachomatis by Conventional PCR
In preceding work, conventional PCR was used to select C. trachomatis-positive carcinoma samples that were later used to establish an in situ PCR assay. Paraffin-embedded material from 20 young women with endocervical carcinomas (10 squamous cell carcinomas in situ, 10 invasive squamous cell carcinomas) and fresh endocervical smears from five patients with and without cellular changes were analyzed for C. trachomatis DNA. By amplifying a 517-
Establishment of a C. trachomatis-specific IS-PCR
The IS-PCR assay established was based on amplification of a 517-

Identification of 517-BP PCR fragments resulting from the cryptic plasmid of C. trachomatis on 3% agarose gel. PCR was used to amplify the DNA fragment from five squamous cell carcinomas (
The best IS-PCR results for smears were generally achieved with specimens that had been fixed with 96% ethanol (v/v) before transport and that contained enough epithelial cells with preserved morphology. Applying the TEN/protease A assay resulted in data that are easy to reproduce. In addition, other digestion techniques were performed, e.g., treating specimens with (a) trypsin (2 mg/ml, 37C) for 20, 30, 40, 50 min and (b) proteinase K (10 μg/ml) for 10 min followed by 0.2% (w/v) glycine for 5 min. However, with these techniques either unspecific background staining or lack of staining was observed.
Detection of C. trachomatis by Electron Microscopy (EM)
To quantify the number of bacteria in an infected cell, the positive control McCoy cells were examined by EM. The cells were embedded in resin and ultrathin sections were cut from the samples. EM indicated that the Lowicryl K4M sections of the McCoy cells contained large numbers of chlamydial inclusion bodies (Figure 3). EM was also performed with cervical smears from a patient with known C. trachomatis infection. In the ultrathin sections, however, the few cells infected could not be detected among other noninfected cells. The paraffin-embedded material was not suited for EM because the morphology of dewaxed cells was easily destroyed by the EM techniques used for embedding.
Specificity of the Direct IS-PCR Assay Applied on Sections
After the IS-PCR procedure and in lieu of staining biotin-labeled PCR fragments, we isolated total DNA from the sections and smears that had shown significant staining of target cells in preceding IS-PCR experiments. In all cases, a weak DNA band of 517
Discussion
Intracellular bacteria represent model organisms for the application of new genetic technologies such as in situ PCR, which allows visualization of amplified target DNA under the light microscope (Long et al. 1992; Komminoth et al. 1992,1993). The aim of our study was to establish an IS-PCR assay for detecting C. trachomatis. Because various authors have referred to a significant association of the infectious agent with intraepithelial neoplasia or invasive squamous cell carcinoma (Paavonen et al. 1979; DeSanjose et al. 1994; Nunez-Troconis 1995), biopsy material from patients bearing one of these tumor entities was selected. Chlamydia-positive histological sections showed strong staining that was localized in the non-neoplastic cylindrical epithelial cells of the endocervical mucosa but not in the carcinoma cells. Signals of comparably strong intensity were observed in infected McCoy fibroblasts, inspiring us to determine the cytoplasmic distribution of bacteria in the positive controls. EM performed on the murine fibroblasts showed that most of the cells were tightly packed with hundreds of inclusion bodies of 0.2–0.4 mm in diameter. Therefore, we presume that at least some of the infected epithelial cells intensely stained by IS-PCR may contain comparably large numbers of inclusion bodies. Notably, some of the infected epithelial cells revealed a strong staining near the basal lamina, indicating a local concentration of bacteria. This might be due to heparan sulfate receptors, which are distributed over the basal cell surface and show a strong affinity for the MOMP protein of C. trachomatis (Wilson et al. 1990; Su et al. 1996). IS-PCR signals were also detected in the non-neoplastic squamous and metaplastic cells of the transformation zone in which the earliest precancerous abnormalities are observed during the histogenesis of cervix cancer. Other working groups investigating cervical swabs by conventional PCR have also referred to the close association between C. trachomatis and the transformation zone (TerMeulen et al. 1995). For this reason, one can speculate that molecular interactions between parasite and host cell contribute to the genesis of tumors. This potential connection between infection and neoplastic process was also confirmed by other viral pathogenic factors in the case of oral cancers (Honig et al. 1995).
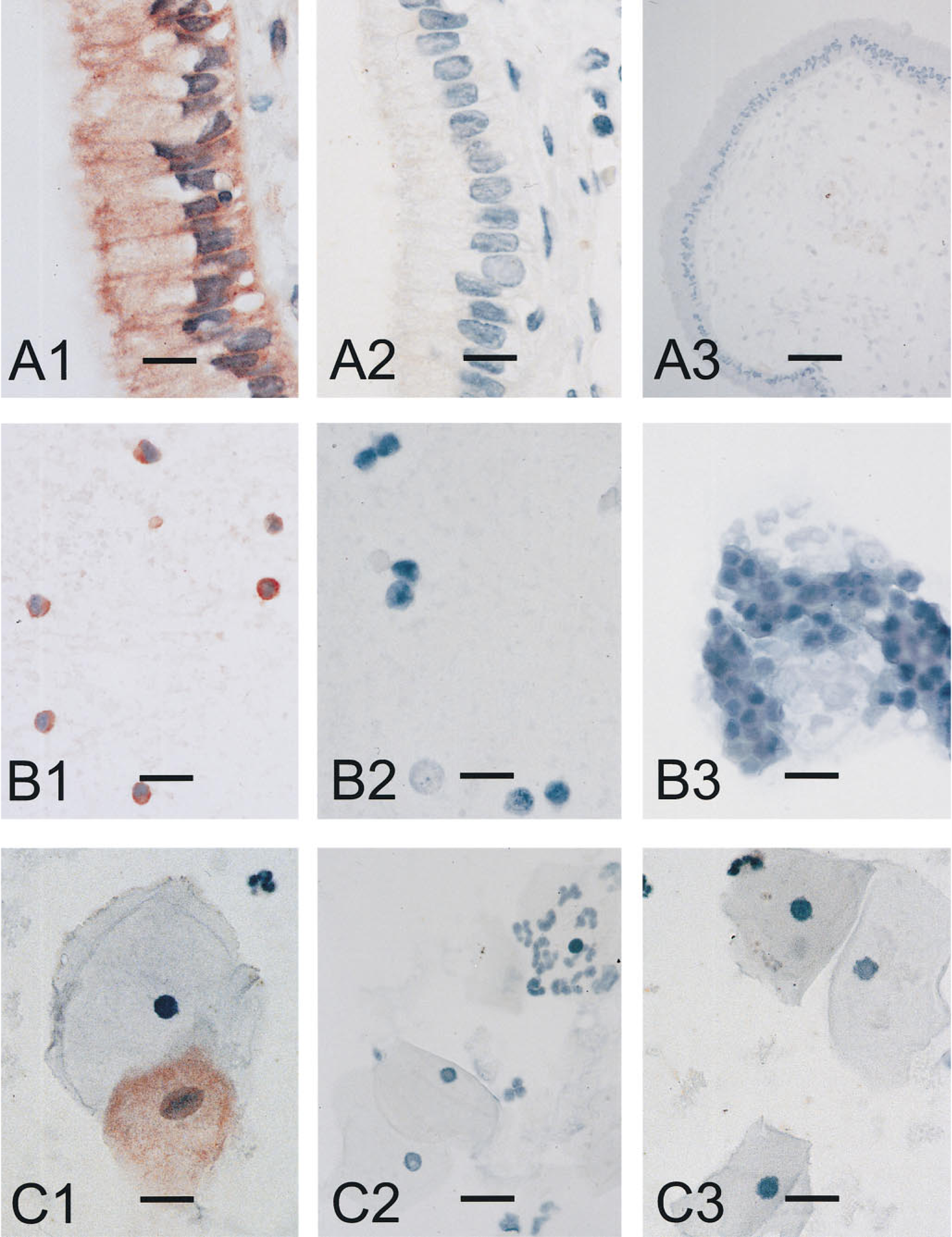
In situ PCR detection of C. trachomatis DNA in endocervical mucosa. The upper row shows columnar epithelial cells of the endocervix from a patient with squamous cell carcinoma in situ. (
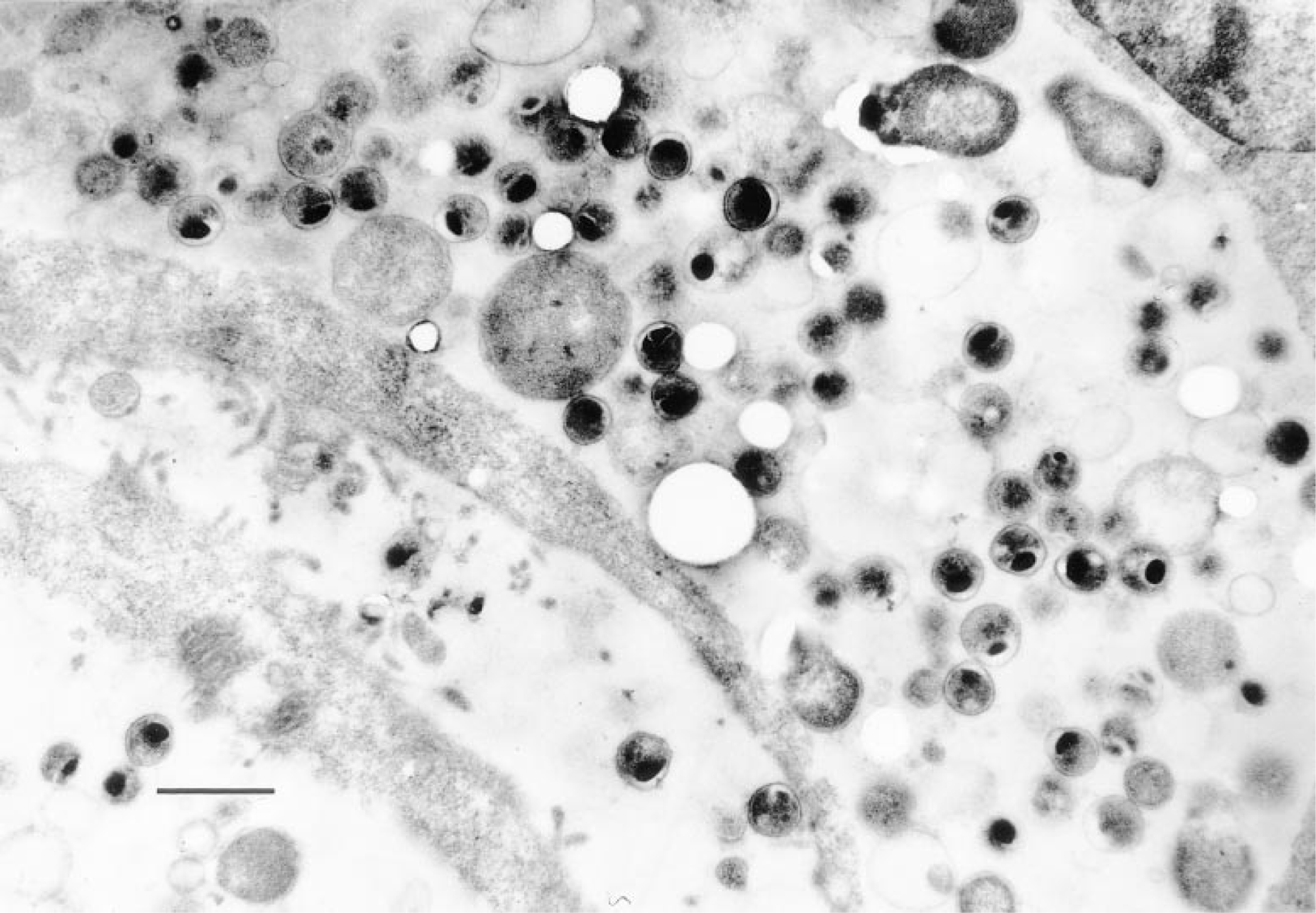
Localization of C. trachomatis in Lowicryl K4M sections of infected cells. The electron micrograph shows a section of a McCoy cell with hundreds of cytoplasmic inclusions characteristic of active chlamydial infection. Mature elementary bodies, reticulate bodies, and intermediate bodies can be seen. Bar = 1 μm.
C. trachomatis infections are difficult to detect in conventional cytological smears because swabs of infected women often reveal normal leukocyte populations and show no inflammatory changes in epithelial cells or only nonspecific cytological features. Such smears contain very low numbers of C. trachomatis bacteria. This is why techniques such as time-consuming cultivation fail, because the transport conditions limit the survival time of the sensitive bacteria outside the human body. Therefore, other genetic techniques, such as sensitive non-IS-PCR and IS-PCR, appear more suited for cytological specimens. In the second part of our study, we used our IS-PCR to localize C. trachomatis in fresh endocervical swabs. We observed fine granular staining of chlamydial DNA in infected cells, which might be attributable to complete proteolytic destruction of the bacterial inclusion bodies. Because the three-layered cell wall of the inclusion bodies most likely consists of protein, as reported for the inclusion membrane of C. psittaci (Rockey et al. 1995), we suspect that protease digestion releases plasmid DNA from the vacuoles. Hence, plasmids floating within the whole cytoplasm of target cells would cause diffuse staining.
Direct IS-PCR is a difficult technique that requires intensive optimization work. The data published to date on this type of IS-PCR technique are still contradictory. Indeed, several working groups have demonstrated that direct IS-PCR on archival histological material leads to false-positive results (Komminoth and Long 1993; Long et al. 1993). Meanwhile, other authors have clearly shown that direct IS-PCR is a reliable technique suited for detection of DNA sequences, such as the p53 gene, in histological material (Martinez et al. 1995). Our results are consistent with the latter report that direct IS-PCR is successful if nonspecific reactions caused by mispriming, DNA repair, or endogenous priming are excluded by proper controls. The reproducibility of PCR signals was guaranteed by the cell specificity of the infectious agent. Because C. trachomatis is restricted to the mucosal surfaces of the cervix, the variety of nonepithelial cell types present in most tissue sections and cervical smears may serve as negative control elements. Furthermore, sequencing of the IS-PCR fragment visualized on agarose gel confirmed that the desired product was indeed amplified.
The C. trachomatis-specific IS-PCR established in this study has important applications. First, this technique enhances the histology- and cytology-based diagnosis of Chlamydia infections because the cell structure is preserved and PCR data can be easily correlated to morphological features. Second, the assay may prove a useful diagnostic adjunct for testing the outcome of antibiotic therapy, an increasingly important factor in light of the fact that chronic infections give rise to nonproliferative as well as antibiotic-resistant forms of bacteria.
Footnotes
Acknowledgements
We thank Dr Marianne Scriba of the Division of Medical Microbiology, Department of Human Genetics and Hygiene, University of Goettingen, for providing us with the C. trachomatis-infected McCoy cells. This study is part of the doctoral dissertation of Cand. Med. Götz Ruda.