
Abstract
Recently, we have established nine nasopharyngeal carcinoma (NPC) cell lines in which only one cell line showed the p53 mutation. For investigation of the p53 mutation in this line, immunostaining using anti-p53 antibody was applied and showed the presence of p53 protein in the cytoplasm but not in the nucleus. Single strand conformation polymorphism analysis of the p53 gene showed one normal and one additional DNA band. Cloning and sequencing of PCR-amplified DNA showed an AGA (arginine) to ACA (threonine) heterozygous point mutation at codon 280. Transfection of the p53 DNA binding sequence and chloramphenicol acetyltransferase assay revealed loss of transcriptional activation function of endogenous p53 protein. Co-localization of the endogenous and the transfected exogenous p53 protein by polyclonal antibodies to anti-p53 protein revealed strong exogenous p53 staining in the transfected nuclei and weak staining of endogenous p53 protein in the cytoplasm. We concluded that (a) a heterozygous point mutation at codon 280 was identified in the NPC-TW 06 cell line; (b) the point mutation may cause the stagnation of mutant p53 protein in the cytoplasm, and loss of its transcriptional activation function; (c) endogenous and exogenous p53 protein can be co-localized at the same time in the transfected cells; and (d) 280 mutant p53 protein in NPC cells does not cause a decrease or increase in sensitivity to chemotherapy.
Keywords
NASOPHARYNGEAL carcinoma (NPC) is one of the most common cancers among Chinese living in Taiwan, South China, Hong Kong, and Singapore (Waterhouse et al. 1982; International Agency for Research on Cancer and International Association of Cancer Registries 1976). Its etiological factors have not been well elucidated. However, NPC has been proposed to be closely associated with Epstein–Barr virus (EBV), mainly because sera of many NPC patients contain a high titer of IgA anti-EBV viral capsid antigen and because many NPC tumor tissues contain EBV DNA according to polymerase chain reaction (PCR) analysis (Brooks et al. 1992; Chang et al. 1990; Raab–Traub et al. 1983; Andersson–Anvret et al. 1977; Klein et al. 1974; Nonoyama et al. 1973). In addition, other factors, such as eating salted fish and Chinese herbs, have also been suspected to be associated with NPC (Yu et al. 1988; Zeng et al. 1983; De The 1982). To investigate the actual relationship between EBV and NPC, we have previously established and characterized nine NPC cell lines (Lin et al. 1990, 1993, 1994) and identified EBV in certain tumor cells in some cell lines and their original biopsy specimens (Lin et al. 1994). Our results suggested that carcinogenesis of NPC in non-EBV-containing tumor cells is not related to EBV infection (Lin et al. 1994). Therefore, to investigate the other possible factors related to NPC tumorigenesis, we have examined expression of some oncogenes in these cell lines, and found that the c-myc oncogene is overexpressed in each cell line to different degrees (Lin et al. 1993). We have also examined expression of the retinoblastoma (Rb) oncosuppressor gene in our cell lines (Lin et al. 1992). The Rb DNA sequence in each cell line shows no rearrangement or deletion, but Rb mRNA and protein expression are variable from cell to cell in each cell line. Because mutation of the p53 gene has been found in many other tumor cells (Caron de Fromentel and Soussi 1992; Hollstein et al. 1991; Levine et al. 1991; Rotter and Prokocimer 1991; Baker et al. 1990; Eliyahu et al. 1989; Finlay et al. 1989) and in a few NPC cases (Sun et al. 1992, 1993; Effert et al. 1992; Spruck et al. 1992), we wanted to know whether our cell lines also contained a mutated p53 gene.
The p53 gene encodes a nuclear protein composed of 393 amino acids, is located on chromosome 17p (region 17p13), and contains 11 exons (the first exon does not encode protein sequence) (Levine et al. 1991). Somatic alterations of the p53 gene have been reported in a spectrum of human cancers, suggesting that this event is required for progress of malignant transformation of many human cells (Caron de Fromentel and Soussi 1992; Hollstein et al. 1991; Levine et al. 1991). Ninety-eight percent of base substitution mutations reported thus far in malignant tumors are clustered between exons 5 and 8 (Caron de Fromentel and Soussi 1992; Hollstein et al. 1991; Levine et al. 1991; Eliyahu et al. 1989; Finlay et al. 1989). The expression of wild-type p53 is able to suppress the transformation of cells by other oncogenes (Eliyahu et al. 1989; Finlay et al. 1989), to inhibit the growth of malignant cells in vitro (Casey et al. 1991; Isaacs et al. 1991; Johnson et al. 1991; Baker et al. 1990; Mercer et al. 1990a, b; Diller et al. 1990), and to suppress the tumorigenic phenotype of transformed cells (Chen et al. 1990). The wild-type allele is quite often lost during tumor development (Levine et al. 1991), and mutant p53 can inactivate the endogenous wild-type protein (Levine et al. 1991) through the formation of oligomers between the wild-type and mutant proteins (Levine et al. 1991; Chen et al. 1990). The mutant p53 proteins are also able to form complexes with a constitutively expressed member of the heat shock protein family, the hsc70 protein (Hinds et al. 1990; Finlay et al. 1988). It has been shown that p53 is a transcriptional activator (Fields and Jang 1990; Raycroft et al. 1990) and therefore can control the cell cycle by activating the genes that suppress cell proliferation. None of the mutants has been found to have transcriptional activity (Kern et al. 1992). p53 protein is also able to inhibit DNA replication (Levine et al. 1991) and induce apoptosis (Clarke et al. 1993).
Most NPC biopsy specimens show a normal p53 gene (Spruck et al. 1992; Sun et al. 1992); only a few of them reveal mutant p53 in primary and metastatic NPC tumor cells (Effert et al. 1992). In NPC cell lines, two lines (CNE1 and CNE2) were reported to have the heterozygous point mutation at codon 280 of p53 (Sun et al. 1992). Recent studies also suggest that p53 status may be an important determinant of tumor response to therapy. For example, it was found that defects in apoptosis due to inactivation of p53 could produce treatment-resistant tumors (Lowe et al. 1994). In this study we used immunohistochemistry, Southern blotting, Northern blotting, PCR, and single strand conformation polymorphism (SSCP) analyses to investigate p53 gene, mRNA, and protein expression in our NPC cell lines. We found that only one cell line (NPC-TW 06) showed an abnormal p53 gene in the original biopsy specimen and in the cell line. The other eight lines revealed a normal p53 gene and protein in their original biopsy specimens but also appeared heterozygous point mutation of p53 protein in the culture cell. Perhaps this kind of mutation in vitro is necessary for their survival in the culture system. Therefore, here we report our results from this line. Some tumor cells in this line were starved and the distribution of their p53 protein investigated. In addition, the starved cells were further incubated in the normal culture medium to see whether it was a reversible phenomenon. To confirm this finding, cycloheximide was also used. Furthermore, the transcriptional activation function of p53 protein in this cell line was observed by chloramphenicol acetyltransferase (CAT) assay. Co-transfection of p53 genes (including wild-type gene and mutant gene) and the reporter gene to NPC cells with the observation of the distribution of the endogenous and exogenous p53 proteins in the transfected cells were performed. We also treated the wt p53 and 280 mt p53 transfectants with cisplatin to observe the response of different p53 status to cisplatin treatment.
Materials and Methods
Cell Cultures
The NPC cell line designated NPC-TW 06 was established and characterized in our laboratory previously (Lin et al. 1993). The tumor cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum (FCS) and antibiotics in a 10% CO2 incubator at 37C. The hepatoma (Hep3B) cell line was obtained from Dr. J.Y. Shew (Department of Biochemistry, College of Medicine, National Taiwan University, Taiwan). The colon adenocar-cinoma cell line (DLD-1) and osteogenic sarcoma cell lines (Saos-2 with deletion of p53 gene and U-2OS with wild-type p53 gene) were obtained from American Type Culture Collection (Rockville, MD) and were grown in DMEM containing 10% FCS.
Immunohistochemical Study
The NPC, U-2OS, and colon adenocarcinoma cell lines were immunostained using a previously described method (Lin et al. 1990). A monoclonal antibody (MAb PAb240) against p53 protein (Oncogene Science; Manhasset, NY) was used. In one additional experiment, we grew cells in DMEM medium containing only 0.2% FCS for 2 days (some cells were reincubated in DMEM containing 5% FCS for another 2 days) and fixed them for immunostaining with PAb240. In another experiment we treated the NPC cells with cycloheximide (10 μg/ml) (Gannon and Lane 1991) for 12 hr and then fixed the cells for immunohistochemistry using polyclonal anti-p53 antibodies. Furthermore, wt p53 transfectant and 280 mt transfectants were treated with cisplatin (5 × 10-5 M) for 24 hr and fixed for immunostaining with the same antibodies. For the negative control, the specific primary antibody was replaced by control ascites obtained from a Balb/c mouse injected IP with a mouse myeloma cell line. [MAb PAb240 recognizes a denaturation-resistant epitope on mutant p53 between amino acids 156 and 335 and does not recognize the wild-type p53 protein (Gannon et al. 1990).] In addition, paraffin sections of the original biopsy specimen from which the NPC-TW 06 cell line was derived were deparaffinized, rehydrated, treated with proteinase K (5 μg/ml) for 10 min, and processed by the above-mentioned staining procedure (Lin et al. 1990). The p53 plasmid-transfected NPC cells were stained by polyclonal antibodies against wild-type p53 protein. The antibodies were obtained by transformation of wild-type p53 plasmid to bacteria for production of a large amount of wild-type p53 protein. The latter was then immunized to rabbit for production of polyclonal antibodies (Hsu et al. 1995).
Southern and Northern Blot Analyses
The isolation of genomic DNA from NPC culture cells was performed as previously described (Lin et al. 1990, 1992). The DNA (10 μg each) was restricted with endonuclease Pvu II and analyzed by 0.6% agarose gel electrophoresis and subsequently transferred to nitrocellulose (NC) membrane for hybridization with a p53 cDNA probe. The membranes were finally exposed to Kodak-X-Omat film (Eastman Kodak; Rochester, NY) for 1–2 days at −70C, using intensifying screens, then developed with an automatic processor. The p53 cDNA was a 1.75-KB DNA fragment and was a gift from Dr. J.Y. Shew. It covered the 11 exons of the p53 gene and was labeled with [32 P]-dCTP by the random priming method (Lin et al. 1990, 1992). Total cellular RNA was extracted from the cultured NPC cell line by the method developed by Birnboim (1988). Total RNA (15 μg/lane) was size-fractionated by electrophoresis in a 1.0% agarose gel containing formaldehyde, then transferred to an NC membrane and probed as described above. A negative control (Hep3B) cell line was also included.
Synthesis of DNA Primers and Oligonucleotides
Oligonucleotide primers were synthesized by the phosphoramidites method using Model 380 Applied Biosystems DNA Synthesizers from the Institute of Biomedical Sciences (IBMS), Academia Sinica, Taipei. After deprotection by ammonia water, the oligonucleotides were purified by affinity column kits. Some primers were purchased from TIB MOLBIL (Berlin, Germany). The oligonucleotide sequence for PCR primers in exons 1–11 and sequencing primers in exons 8–9 were used according to published materials (Lehman et al. 1991).
PCR Analysis
Genomic DNA (∼500 ng) was amplified by PCR using both p53-specific primers (Lehman et al. 1991) in the intron regions surrounding exons 1–11. PCR reaction was performed according to a published method (Lin et al. 1994). The PCR products were checked by gel electrophoresis.
SSCP Analysis
The procedure for SSCP was performed according to a published protocol (Ausubel et al. 1988). The sense or anti-sense primer was 5′ end-labeled with [32 P]-dATP by T4 polynucleotide kinase before the PCR program proceeded. The PCR products were denatured for 5 min at 98C in a solution containing 90% formamide, 20 mM EDTA (pH 8.0), 0.5% xylenecyanol, and bromophenol blue, then chilled to 4C. The denatured PCR products were separated by 6% native polyacrylamide gel electrophoresis at room temperature for 3–6 hr at 45 watts. The gels were dried under vacuum and exposed to X-ray film with intensifying screens at room temperature.
Clone Sequencing
Genomic DNA (0.5 μg) prepared from the NPC cell line was used in 100 μl PCR reaction mixture. The DNA cloning sequences at exon 8–9 for PCR primers were 5′-GGGAAG-CTTTTGGGAGTAGATGGAGCCT-3′ and 5′-GGGGAA-TTCAGTGTTAGACTGGAAACTT-3′. The PCR products were digested with EcoRI and HindIII, fractionated by 2% agarose gel, and specific bands electroeluted. The purified products were ligated into an EcoRI-HindIII-cleaved pGEM3 vector (Promega; Madison, WI). After bacterial transformation, at least four different p53 exon 8–9 subclones from the cell line were selected for sequencing. For comparison, normal control DNA from human peripheral blood cells was also included.
Two approaches were used to determine the DNA sequences of selected clones. The first approach was by direct sequencing of the amplified DNA via Sanger's dideoxy method using one of the sequencing primers (Ausubel et al. 1988). Briefly, purifed DNA (PCR product), 1–2 μg was denatured at 98C for 3 min, annealed with 3 pmol sequencing primer, and sequenced with the T7 Sequenase kit reagents (Pharmacia; Uppsala, Sweden). Radioactive label incorporation was achieved with incubation at room temperature for 5 min with 35 S-labeled deoxynucleotide (New England Nuclear; Boston, MA) corresponding to the first base of the nascent chain. Samples were run on 8% urea polyacrylamide gels (Gel-Mix 8; Gibco BRL, Gaithersburg, MD) for 2–5 hr. Dried gels were placed against Kodak X-AR-5 film at room temperature for 1–2 days. The other approach was automatic DNA sequencing by the sequencing unit in the Institute of Biomedical Sciences (IBMS), Academia Sinica, Taipei.
The DNA sequences obtained were then compared with published p53 sequences for sequence homology using the sequence analysis software package of the Genetics Computer Group (Version 6.0, 1989; University of Wisconsin Biotechnology Center, Madison, WI).
Construction of Reporter Plasmid 3PBS-CAT
The palindromic oligonucleotide of p53 consensus DNA binding sequence (5′-AGCTAGGCATGTCTAGACATGCCT-3′) (El-Deiry et al. 1993), synthesized in IBMS, was phosphorylated with T4 polynucleotide kinase. After purification twice by isopropanol precipitation, the oligonucleotide was boiled for 5 min and kept in a water bath to cool slowly. The self-annealed oligonucleotides were cloned into the HindIII restriction site of the E1b-CAT plasmid with T4 DNA ligase. After bacterial transformation, the plasmid DNA was extracted to screen the clones which contained three copies of p53 consensus DNA binding sequence (3PBS-CAT). The E1b-CAT plasmid, consisting of a chloramphenicol acetyl-transferase gene in Psp 72 plasmid, was obtained from Dr. Y.S. Lin (Hsu et al. 1995) of IBMS.
Construction of Plasmids Containing Wild and Mutant p53 Genes
The wild-type and mutant p53 gene cloning from our NPC cell line was performed using methods similar to those mentioned above. The primers for cloning of NPC-TW 06 p53 wild-type and mutant type genes were 5′-GGGAAGCTTGCCGCCATGCATCATCATCATCATCATATGGAGGAG-CCGCAGT-3′ and 5′-GGGGGATCCTCAGTCTGAGTCAGGCCC-3′, which included the whole coding sequence (1.3 KB). The PCR products of cDNA were inserted into the pCEP4 vector, which contained CMV IE gene as the promoter.
Transfection
Using the calcium phosphate procedure (Di Nocera and Dawid 1983), the reporter plasmids alone or in combination with pCEP4-p53 constructs were transfected into U-2OS, Saos-2, and NPC cell lines. Similarly, the pCEP4-wt p53 or 280 mt-p53 constructs were also transfected to NPC cell lines, respectively. Briefly, each cell line was grown at 75% confluence in 10-cm plates. Aliquots of 0.5 ml of 0.25 M CaCl2, containing 5 μg of purified plasmid DNA, were added dropwise to an equal volume of 2 × HeBs [2 × HeBs contains (in g/liter): NaCl 16; KCl 0.7; Na2HPO4 0.4; dextrose 2; Hepes 10, at pH 7.1] in a 35-mm tissue culture dish and was gently rocked by hand. After 30 min at room temperature, the suspension of calcium-phosphate complexes was gently pipetted into the cell culture dishes, which contained 10 ml of culture medium. The cultures were left undisturbed until harvest 2 days later. Stable clones of the NPC-TW 06 expressing wt-p53 or mt-p53 were selected in the medium containing 5 U/ml of hygromycin B (Sigma; St Louis, MO). Hygromycin B resistant clones were cloned and subsequently expanded for other experimental analyses.
CAT Assay
Cells transfected with different plasmids were harvested, washed twice in PBS, and resuspended in 100 μl of 0.5% Triton X-100, 0.25 M Tris-HCl (pH 8.0) at 4C, for 2 min. After the cells were spun for 5 min in an Eppendorf microfuge at 4C, the supernatants were removed and assayed for enzyme activity (Gorman et al. 1982). The assay mixture contained (in a final volume of 180 μl) 100 μl of 0.25 M Tris-HCl (pH 7.5), 4 μl of 80% glycerol, 30 μl of cell extract, 1 μCi of [14C]-chloramphenicol (50 μCi/mmol; Amersham; Poole, UK), and 5 μl of 4 mM acetyl coenzyme A. After incubation for 2 hr at 37C, the reaction was stopped with 500 μl of cold ethyl acetate; the latter was also used to extract the chloramphenicol. The organic layer was dried and dissolved in 10 μl of ethyl acetate, spotted on silica gel thinlayer plates, and run with chloroform–methanol (95:5). The plates were finally exposed to Kodak-X-Omat film for 1–2 days at −70C, using intensifying screens, and developed in an automatic processor.
Assay for Cell Viability in wt-p53 and 280 mt-p53 Transfected NPC Cells After Cisplatin Treatment
Cell viability was assessed by MTT [3-(4, 4-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide; Sigma] assay (Carmichael et al. 1987). One day before the experiment, cells were plated in 96-well tissue culture plates at a density of 2–4 × 103 cells/well. After treatment with cisplatin for various periods, cells were incubated in MTT solution (0.1 mg/ml) for 2 hr at 37C, and absorbance was measured using a model 550 microplate reader with wavelength of 540 nm. Cell viability was defined as follows:
cell viability = (experimental value − X)/(control value − X)
where × was the value obtained in wells containing medium and MTT without cells.
The relative survival rate of wt-p53 transfectant to survival rate of mt-p53 transfectant after cisplatin treatment was obtained compared with the survival rate of non-transfected NPC-TW 06 cells after cisplatin treatment.
Results
Immunolocalization of p53 in the NPC Cell Line and Its Original Biopsy Specimen
When we used an MAb against mutant p53 (PAb240) to stain our NPC cell line, reaction product of anti-p53 was seen in the cytoplasm but not in the nucleus of each single cell (Figure 1A). Some cells revealed strong cytoplasmic staining, whereas others showed rather weak staining. All mitotic cells showed strong cytoplasmic but not chromosomal staining (data not shown). NPC cells starved in culture medium containing 0.2% FCS for 2 days showed nuclear but not cytoplasmic staining (Figure 1B). When the original culture medium containing fetal calf serum was used to reincubate with these starved cells for another 2 days, the p53 protein was localized in the cytoplasm (data not shown). When the cells were treated with cycloheximide, the p53 protein was localized to the nucleus (Figure 1C). If NPC cells were treated with cisplatin (5 × 10-5 M) for 1 day, the p53 protein was still localized in the cytoplasm (Figure 1D). When the original biopsy specimen from which the cell line was established was stained by the same antibody, the paraffin section (NPC-TW 06) revealed the same pattern of cytoplasmic but not nuclear staining in some tumor cells (Figure 2). Other tumor cells in the same specimen were not stained. When the positive control cells (colon adenocarcinoma) were stained with PAb240, both cytoplasmic and nuclear staining could be identified. If nonspecific ascites was used to stain the NPC cell line and colon cancer cell line, none of the tumor cells was specifically stained (data not shown).
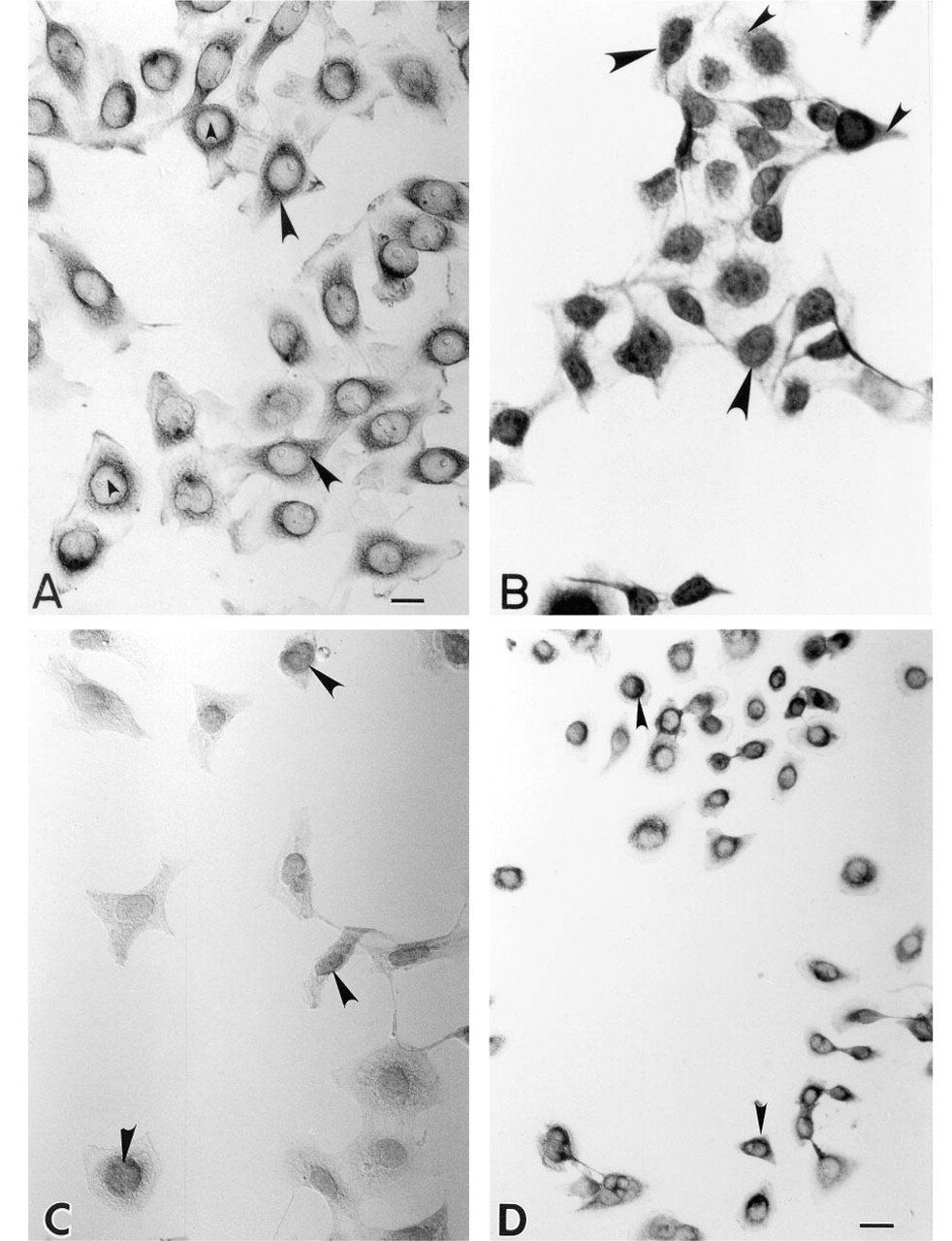
Immunohistochemical localization of p53 protein in NPC cell lines with or without cycloheximide and cisplatin treatment.
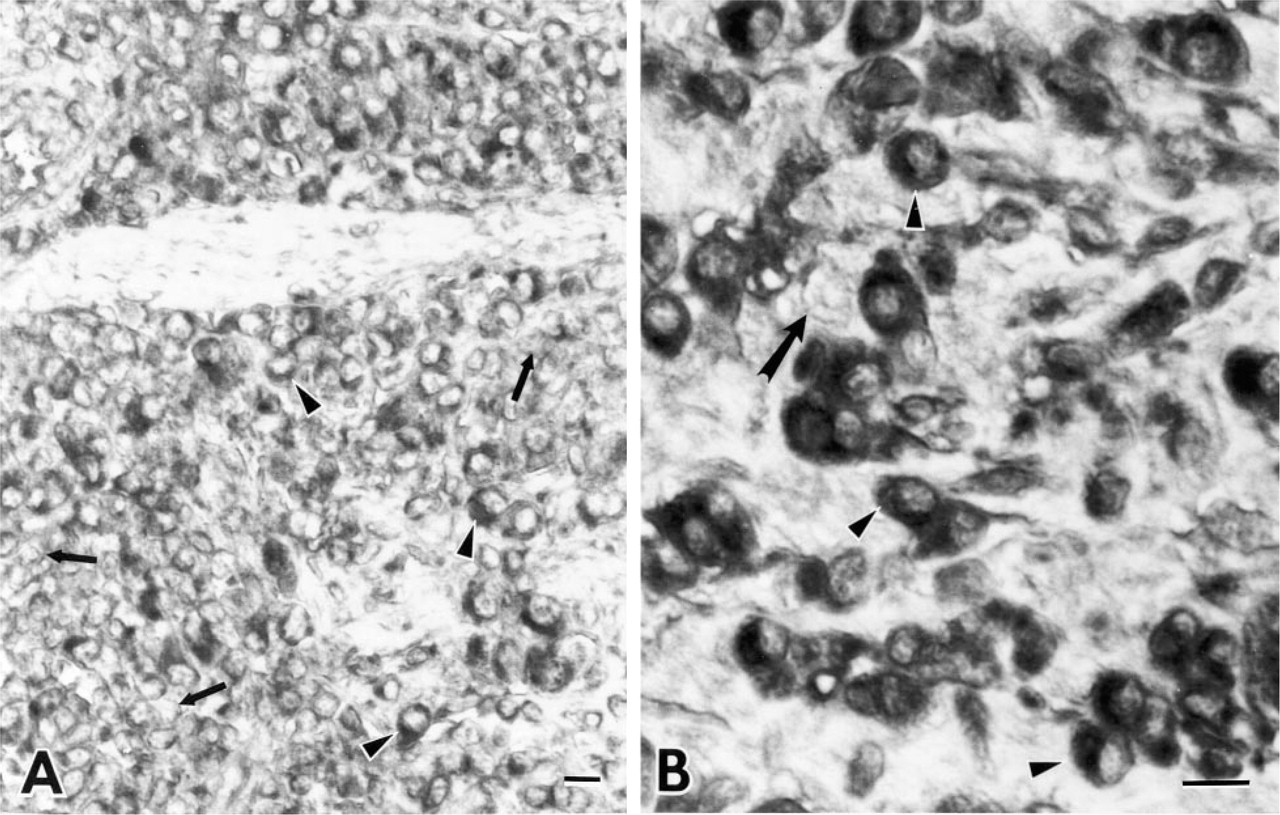
Immunohistochemical localization of p53 protein in the original biopsy specimen of NPC-TW 06 cell line. Reaction product of PAb240 is shown in the cytoplasm but not in the nuclei of some tumor cells (arrowheads). Other cells are not stained (arrows).
Southern and Northern Blot Analyses of p53 DNAs and RNAs in the NPC Cell Line
When the NPC high molecular weight DNA was digested with PvuII and analyzed by a p53 DNA probe, three bands of 10.5, 3.9, and 1.6 KB were identified in the NPC cell line and in the positive control DNA from normal human peripheral blood cells. A negative control cell line (hepatoma Hep3B) revealed only two bands, of 10.5 and 1.6 KB (data not shown). In Northern blot analysis, the NPC cell line showed a p53 band, whereas the negative control hepatoma Hep3B cell line revealed no p53 band (data not shown).
PCR and SSCP Analyses of the p53 Gene in the NPC Cells
In the gel analysis of PCR-amplified products from 11 exons, no specific abnormal band could be identified compared with p53 products from normal human peripheral blood cells (NHB) (data not shown). However, by SSCP, two distinct bands, one moving faster (F band) and the other more slowly (S band), could be clearly identified. The density of the F band was stronger than that of the S band. The mobility of the S band in the NPC line was similar to that of the normal control DNA (NHB cells) (Figure 3).
Cloning and Sequencing
After the pGEM3 vectors, which had been inserted with our PCR products from the exon 8–9 region, had been transfected into the E. coli MC1061, four clones of bacteria from the culture plates were selected and amplified. The amplified DNA fragments were sequenced by both direct and automatic sequencing. Direct sequencing from some clones showed a point mutation at codon 280 from AGA to ACA (Arg→Thr) (Figure 4B), whereas some other clones revealed normal sequence (Figure 4C), similar to the normal control DNA sequence (Figure 4A). The automatic sequencing showed the same results.
CAT Assay
The p53 consensus binding sequence was cloned into the CAT plasmid, then transfected into the NPC cells, U-2OS cells, and Saos-2 cells. The CAT activity from each transfected cell line was then examined by thin-layer chromatography. Results showed that the NPC cell line (Figure 5, Lane 3) and the Saos-2 cell line (Figure 5, Lane 2) contained no CAT activity, whereas the U-2OS cell line (Figure 5, Lanes 1 and 5) had clear CAT activity. When both 3PBS-CAT plasmids plus pCEP4-wt p53 (pCEP4-wild type p53) or plus pCEP4-mt 280p53 (pCEP4-mutant type p53) were co-transfected into Saos-2 cells, CAT activity was seen only in the cells transfected with wt p53 plasmid (Figure 6A, Lane 1), and not in the cells transfected with mutant 280 p53 plasmid (Figure 6A, Lane 2). When 3PBS-CAT plasmid and wild-type p53-pCEP4 plasmid were co-transfected to the NPC cells, rather weak CAT activity was present (Figure 6B, Lane 1) compared with that in 3PBS-CAT-transfected U-2OS cells (Figure 6B, Lane 4). However, when the mt 280 p53-pCEP4 plasmid was used to replace wild-type p53-pCEP4 plasmid, no CAT activity could be seen in the NPC cells (Figure 6B, Lane 2). A summary of CAT assay results is shown in Table 1. The transfection efficiency of each constructed plasmid to the cells was judged by the immunostaining of p53 protein in the transfected cells (see below).
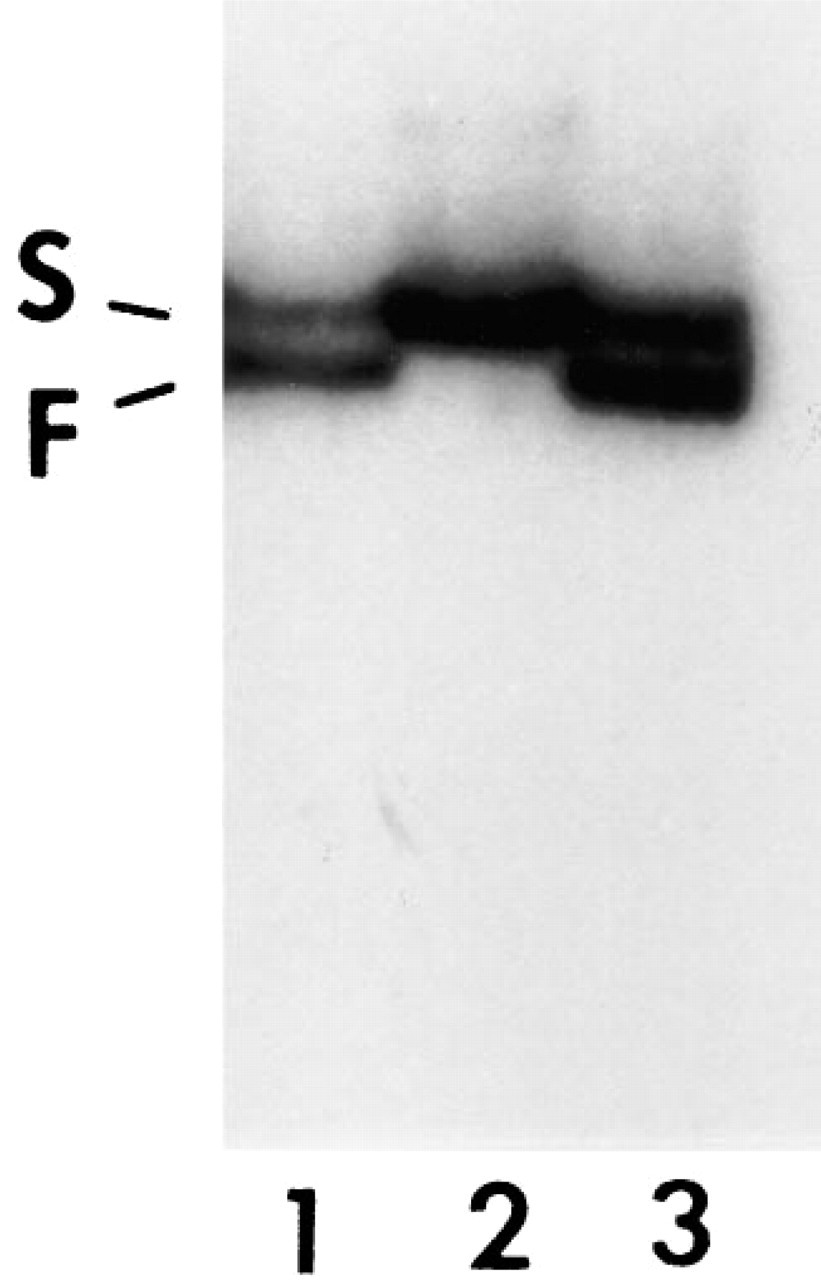
Single strand conformation polymorphism analysis of p53 gene in an NPC cell line. Two distinct bands, one that moved faster (F band) and the other more slowly (S band), are seen in the cell line (Lanes 1 and 3); the intensity of the F band is stronger than that of S band. The normal control (normal human peripheral blood cell DNA) (Lane 2) shows one strong S band only. Lane 3 contained a higher concentration of PCR product than Lane 1.
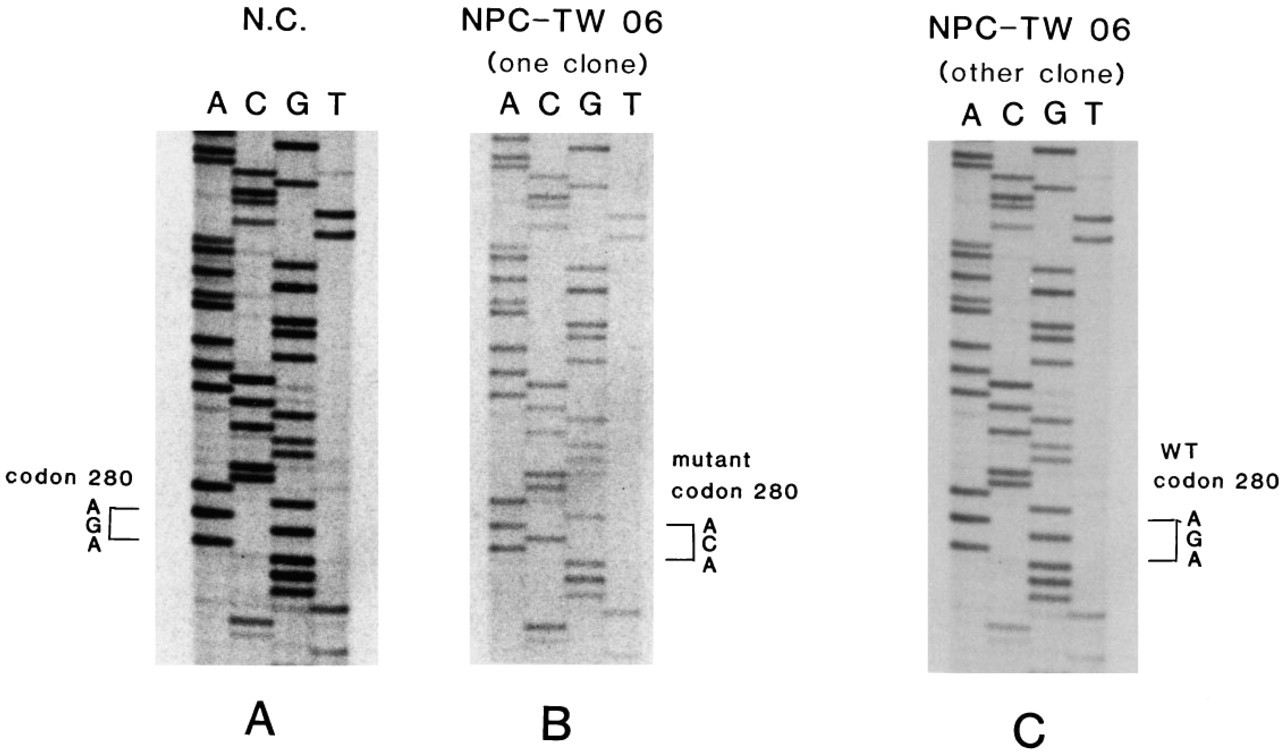
Direct DNA cloning sequencing of p53 gene at exon 8 in the NPC-TW 06 cell line. One point mutation from G to C at codon 280 is shown in one mutant clone p53 gene
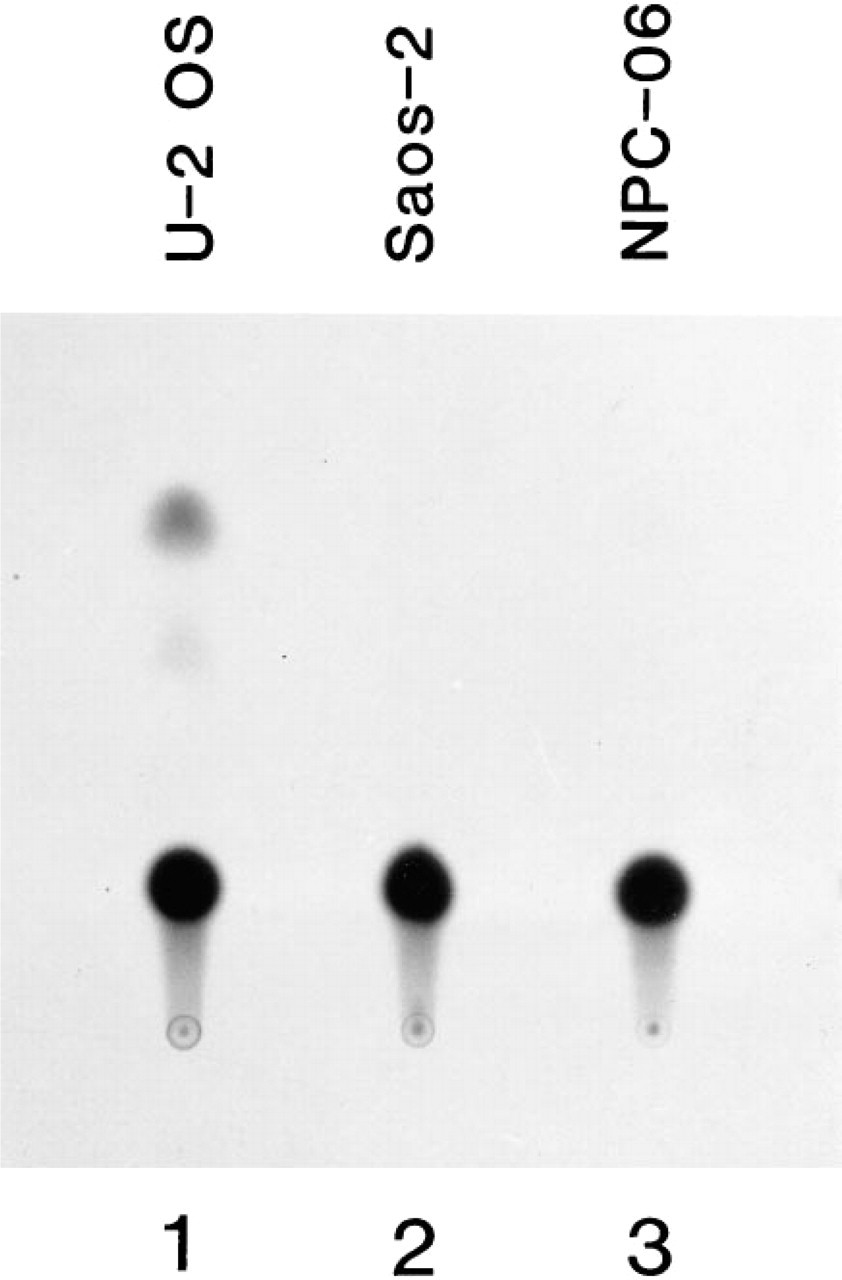
Chloramphenicol acetyltransferase (CAT) assay of transcriptional activation function of endogenous p53 protein in NPC cell lines. p53 DNA binding sequence-containing CAT plasmid (3PBS-CAT) was transfected into NPC cell lines and into Saos-2 and U-2OS cell lines (an osteosarcoma cell line containing normal p53 gene). Lane 1, U-2OS cell line showing positive CAT activity; Lane 2, Saos-2 cell line revealing no CAT activity; Lane 3, NPC-06 (NPC-TW 06) cell line showing no CAT activity.
Identification of Wild-type (wt) and Mutant (mt) 280 p53 Protein in Saos-2 and NPC Cells
When the Saos-2 cells were transfected with a wild-type p53 gene and the expression of normal p53 protein in the cells was detected by immunohistochemical localization, reaction product of anti-wt-p53 protein was present mostly in the nucleus and very weakly in the cytoplasm. The cells containing strong nuclear staining always showed a rounded appearance. When the cells were cultured for several days, those rounded cells revealed degenerative change. When the mt 280 p53 gene was transfected, the distribution of mt 280-p53 protein in the Saos-2 cells was the same as that of the wt-p53 protein. Even when a one-to-one ratio of wt-p53 and mt 280-p53 genes was transfected, the distribution of p53 was similar to that of individual transfections of wt-p53 or mt 280-p53 gene (data not shown). When NPC cells were transfected with the wt-p53 gene, the p53 protein could be observed both in the nuclei and cytoplasm of those cells containing the exogenous wt-p53 gene, with some cells containing strong nuclear and cytoplasmic staining and other cells with strong nuclear and weak cytoplasmic staining. The cells containing strong nuclear and cytoplasmic staining also showed a rounded morphology. When they were cultured for more than 3 days, most of them exhibited degenerative change. In contrast, the untransfected cells displayed only weak cytoplasmic staining (Figure 7A, small arrows). When the mt 280-p53 gene was transfected, a similar staining pattern was seen in the wt-p53 gene-transfected NPC cells (Figure 7B), except that the degenerative cells were fewer than those of the wt-p53 gene transfection). A summary of p53 protein distribution in the Saos-2 and NPC cells is given in Table 2. The number of strongly nuclear-stained tumor cells was about 40–60% of the total cell population in each experiment, indicating that the transfection efficiency in our experiment was about 50%.
Assay for Cell Viability in wt-p53 and 280 mt-p53-transfected NPC Cells after Cisplatin Treatment
The IC50 of cisplatin to NPC cells was 5 × 10-5 M. When wt-p53 transfectants and nontransfected NPC cells were treated with IC50 of cisplatin for 24 hr, a ratio of 0.66 was identified by the MTT assay. Similarly, if the 280 mt-p53 transfectants were used to replace the wt-p53 transfectants, a ratio of 0.70 was obtained.
Discussion
Immunolocalization of p53 protein in many tumors has been reported previously (Bosari et al. 1995; Stenmark Askmalm et al. 1994; Bartek et al. 1993; Moll et al. 1992, 1995; Mayall et al. 1992; Porter et al. 1992; Gannon and Lane 1991). Most reports have shown nuclear localization of p53 protein, whereas only a few have revealed its cytoplasmic localization (Bosari et al. 1995; Stenmark Askmalm et al. 1994; Moll et al. 1992, 1995; Gannon and Lane 1991). Here we have clearly demonstrated that p53 protein was present mostly in the cytoplasm but very rarely in the nucleus of one of our NPC cell lines. Because stagnation of p53 protein in the cytoplasm may be due to a result of abnormal nuclear localization signal or to other factors, such as a cytoplasmic factor that binds to the mutant p53 protein, resulting in the cytoplasmic localization of p53 protein, we performed an experiment to answer this question. When NPC cells were markedly deprived of nutrients for 2 days, the stagnation of p53 protein in the cytoplasm disappeared, and all p53 protein became localized in the nucleus. This phenomenon was further confirmed by an experiment using cycloheximide to block protein synthesis. However, this phenomenon was reversible when the normal culture medium was reused or the cycloheximide was removed, indicating that the NPC p53 protein is an abnormal protein that can not be transported into the nucleus under physiological conditions but becomes a transportable protein and can move into the nucleus under conditions of decreased synthesis of cellular protein. These findings also suggest that some shortlived protein factor(s), which may bind to the mutant p53 and anchor it in the cytoplasm under normal physiological conditions, are unable to anchor the mutant p53 protein under starvation conditions. The p53 protein, when stagnated in the cytoplasm, cannot activate its target genes in the nucleus. Similarly, a recent article (Gannon and Lane 1991) reports that rat cells transformed by a mutant p53 (p53val1 35) and a ras oncogene showed cytoplasmic localization of mutant p53 at 37C, but the protein moved to the nucleus at 32C. Inhibition of protein synthesis by cycloheximide revealed the same effect as in our starvation experiment. The authors concluded that the presence of a short-lived protein is responsible for holding p53 in the cytoplasm at 37C but not at 32C (Gannon and Lane 1991).
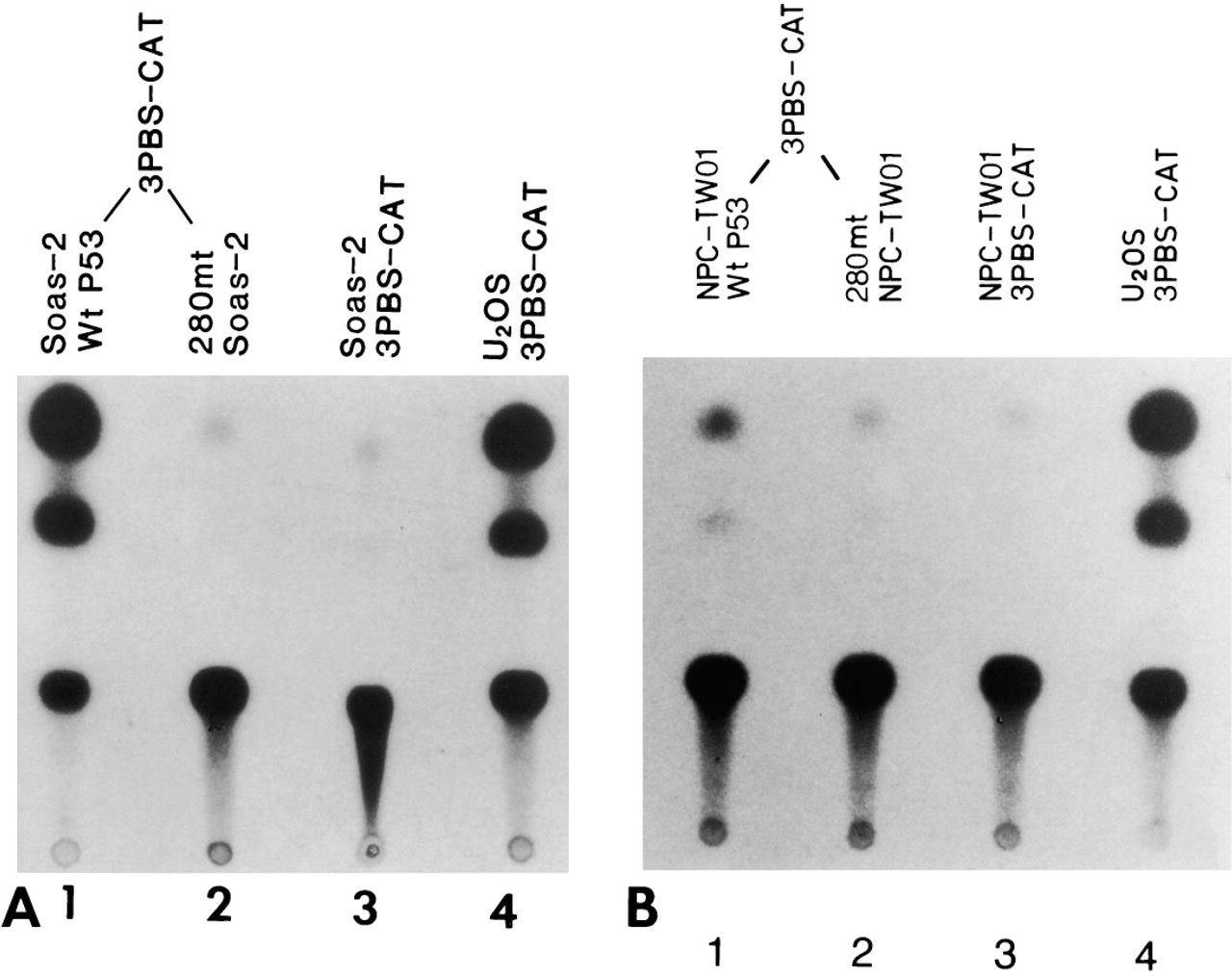
Analysis of wild-type and mutated p53 gene function in Saos-2 cell line and NPC-TW 06 cell line. pCEP4-wt p53 and 3PBS-CAT were co-transfected into Saos-2 cells (
Summary of chloramphenicol acetyltransferase activity in specific DNA plasmid-transfected cell lines a
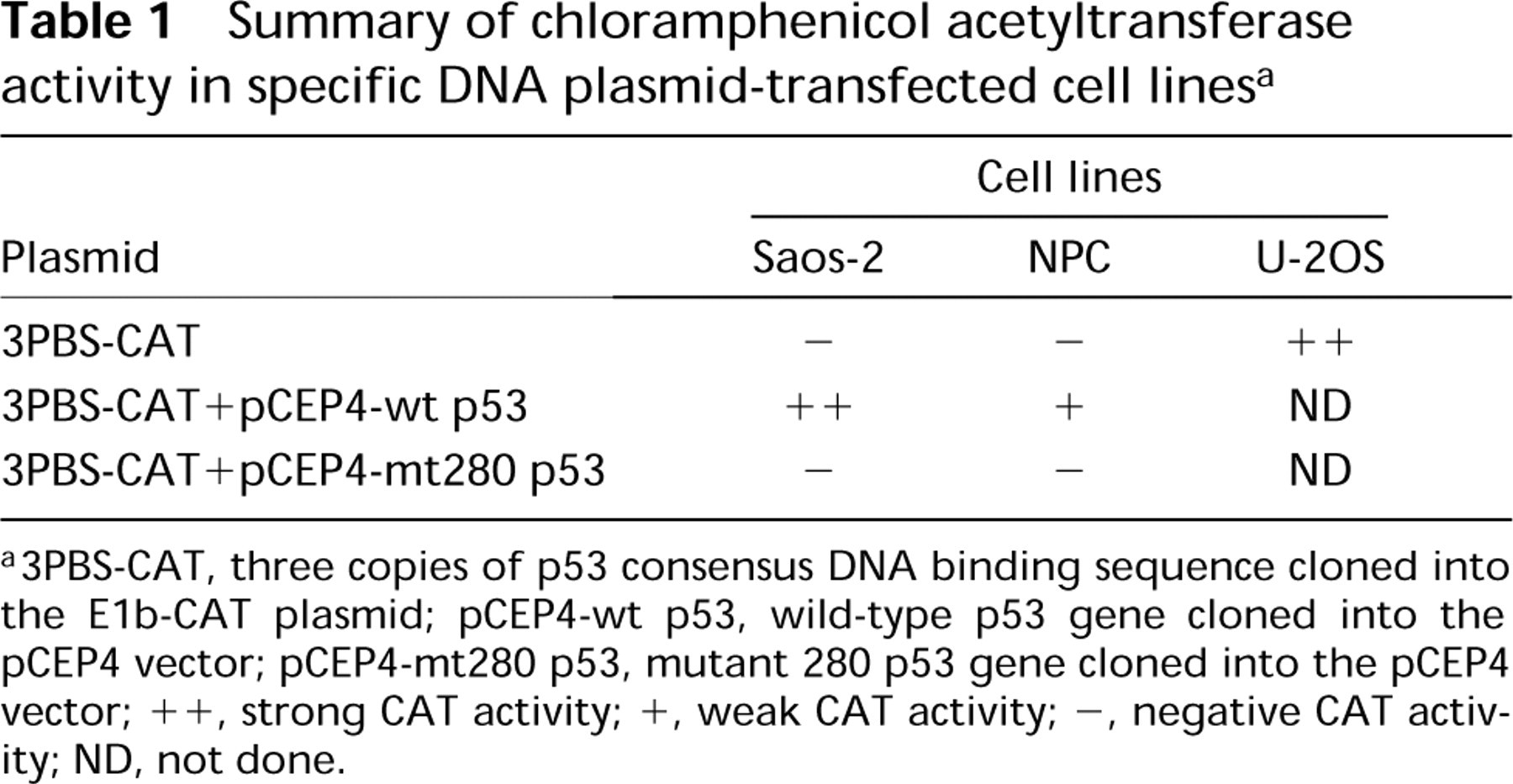

a3PBS-CAT, three copies of p53 consensus DNA binding sequence cloned into the E1b-CAT plasmid; pCEP4-wt p53, wild-type p53 gene cloned into the pCEP4 vector; pCEP4-mt280 p53, mutant 280 p53 gene cloned into the pCEP4 vector; ++, strong CAT activity; +, weak CAT activity; −, negative CAT activity; ND, not done.
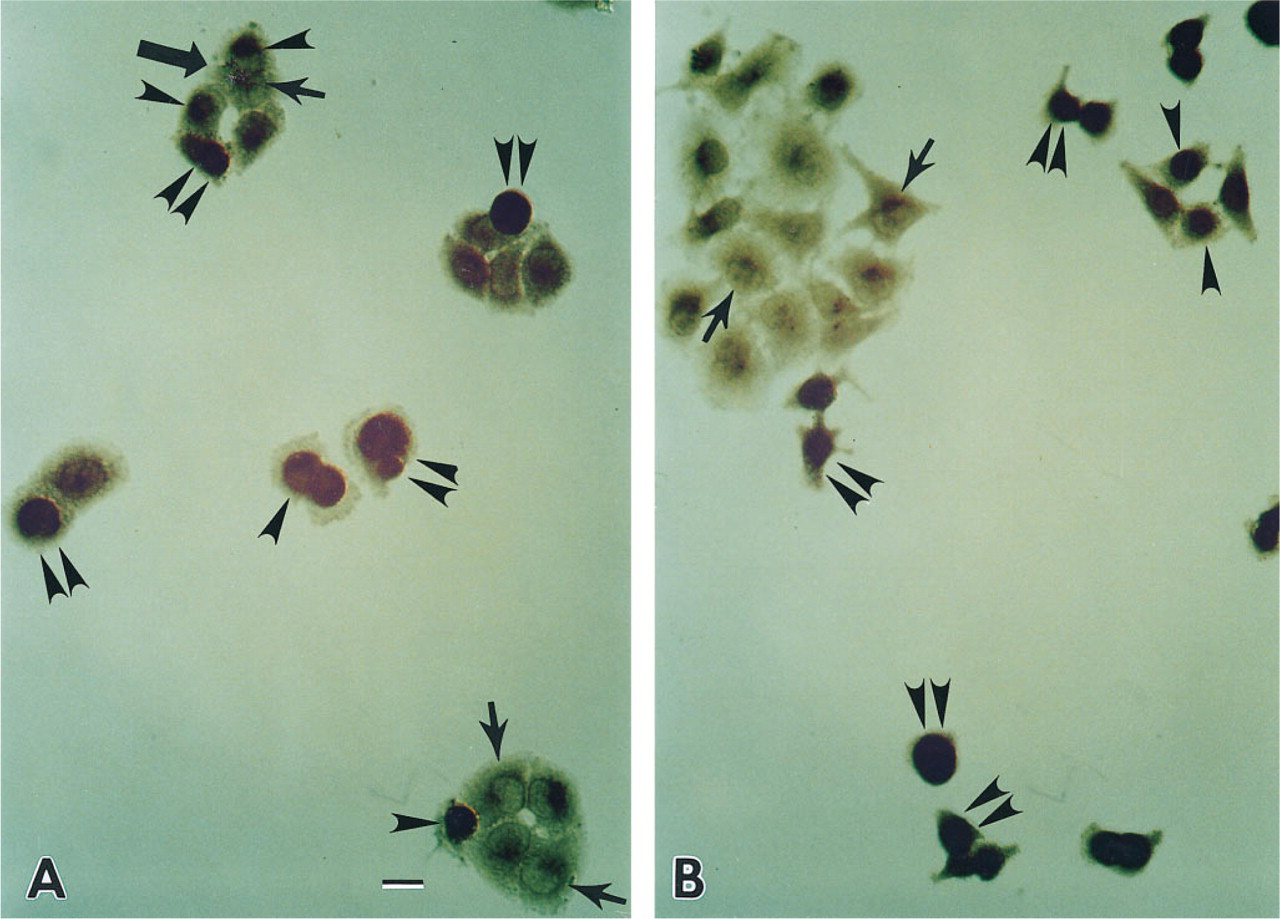
Identification of wild-type and mutant 280 p53 proteins in NPC cells by immunohistochemistry using polyclonal antibodies against wild-type p53.
The abnormality of p53 protein in NPC cell line does not necessarily mean that it is also abnormal in a patient's tumor tissue. To clarify this situation, we immunostained sections of the paraffin block from which our NPC cell line had been established. The sections revealed similar cytoplasmic but not nuclear staining in certain tumor cells, but other tumor cells in the same section showed no p53 reaction product. These findings suggest that the NPC-TW 06 patient's tumor tissue may contain two types of tumor cells: some cells with a mutated p53 gene and other cells with a wild-type p53 gene. They also suggest that the mutation of the p53 gene in these NPC cells does not occur in the initiation stage of tumorigenesis.
Summary of localization of exogenous and endogenous p53 protein in Saos-2 and NPC cells a
a3PBS-CAT, three copies of p53 consensus DNA binding sequence cloned into the E1b-CAT plasmid; pCEP4-wt p53, wild-type p53 gene cloned into the pCEP4 vector; pCEP4-mt280 p53, mutant 280 p53 gene cloned into the pCEP4 vector; ++, strong CAT activity; +, weak CAT activity; −, negative CAT activity; ND, not done.
Our results from Southern blotting to investigate whether the p53 gene was abnormal suggest that no specific gross rearrangement or deletion of the p53 gene exists in this NPC cell line. Similarly, Northern blot analysis also indicates that transcription of the p53 gene is unproblematic, unlike that in the osteogenic sarcoma (Saos-2) and hepatoma (Hep3B) cell lines, which showed notable p53 deletion (Chandar et al. 1992; Bressac et al. 1990). We also performed Western blotting and showed that the p53 protein in this NPC cell line has a normal molecular size (data not shown). The above experiments suggest that the abnormal localization of p53 protein is probably due to specific point mutation. We therefore used both PCR and SSCP to identify the mutated region in the cell line. PCR analysis revealed normal DNA size in each region. However, SSCP showed an additional abnormal band in exon 8 in this NPC cell line (the other exon regions were normal). The finding that the p53 protein was localized only in the cytoplasm in the cell line and that SSCP revealed one normal and one abnormal band (with the density of the F band stronger than that of the S band) in the NPC cell line suggests that our NPC cell line may contain two subpopulations of culture cells, i.e., one population contains a heterogeneous p53 gene having a normal allele and a mutated allele in each cell, and the other subpopulation may contain a homozygous mutated p53 DNA sequence. To identify the precise point mutation, we selected four clones from the cell line for both direct manual and automatic sequencing. Results showed that some clones revealed a point mutation at 280 codon from G to C (Arg→Thr); other clones appeared to have a normal p53 sequence. The finding that the defect of nuclear transport of p53 protein appeared in this cell line may be due to this particular mutation of G to C at codon 280. This mutant p53 may induce a conformational change of p53 protein, resulting in formation of a protein complex with other cellular protein factor(s) in NPC cells, and may be held in the cytoplasm as mentioned above.
The wild-type p53 protein can form a tetramer with a mutant p53 protein (Friedman et al. 1993; Sturzbecher et al. 1992). Because our cloning sequencing indicates that both wild-type and mutant p53 alleles appear in certain populations of tumor cells, it is possible that each of these population of cells synthesizes both the wild-type and the mutant p53 proteins. After both types of p53 protein are synthesized, they may form tetramer proteins which, in turn, can be anchored by the cellular protein factor(s) through the binding of cellular protein factor(s) and p53 tetramer protein. Thus, mutant p53 protein would prevent the wild-type p53 protein from migrating into the nucleus, resulting in retention of p53 tetramer protein in the cytoplasm. In the case of homozygous mutant p53 protein, the latter may be bound by the cellular protein factor(s) directly.
To be sure that most wild-type p53 protein molecules have been trapped in the cytoplasm, resulting in loss of p53 protein in the nucleus to perform its physiological function, we transfected a CAT plasmid construct containing a p53 DNA binding sequence into our NPC and Saos-2 (with deletion of p53 gene) cells. No CAT activity was detected in our NPC and Saos-2 cell lines, in contrast to the positive control cell lines U-2OS (with normal p53 gene), in which the CAT activity was easily detected. This result strongly indicates that, in NPC cells, neither wild-type p53 protein nor mutant p53 protein could get into the nucleus to activate its target genes. To determine whether the mutant p53 gene product in the NPC cell line had lost its transcriptional activation function, we co-transfected mutant p53-pCEP4 plasmid and 3PBS-CAT reporter plasmid into the NPC cells, which resulted in no CAT activity. However, NPC cells that had been transfected with mutant 280 p53 gene showed strong exogenous mutant p53 protein in their nuclei, suggesting that although the excess exogenous mutant p53 protein can not totally be bound by the limited cytoplasmic factor(s) and can move into the nuclei, this protein still can not activate the CAT gene to synthesize enzyme. Similarly, co-transfection of mutant p53-pCEP4 plasmid and 3PBS-CAT reporter plasmid into the Saos-2 cells also showed no activity of CAT enzyme, whereas the exogenous mutant p53 protein could also be detected in the nuclei of transfected Saos-2 cells. Both experiments strongly indicate that our NPC mutated p53 protein has already lost its transcriptional activation function.
To evaluate NPC cellular factors that may play a role in trapping the mutant and wild-type p53 protein complex, we also co-transfected wild-type p53-pCEP4 and 3PBS-CAT plasmids into both Saos-2 and NPC cells. Our results, showing that co-transfection of wild-type p53 and 3PBS-CAT into the p53-deleted Saos-2 cells exhibited strong CAT activity, whereas co-transfection into the NPC cells revealed weak CAT activity, suggest that some wild-type p53 protein may also be trapped in the NPC cytoplasm and that only part of its excess protein which is not bound by the saturated endogenous mutant 280 p53 protein can get into the nucleus to activate the CAT gene. This interpretation is supported by the results from immunostaining of the transfected cells, which showed strong exogenous mutant p53 protein in both the nuclei and cytoplasm in some cells and strong staining in the nuclei and weak in perinuclear region in other cells (Figure 7A). The findings that most wild-type p53 gene-transfected Saos-2 and NPC cells showed fewer cytoplasmic processes with smaller cell bodies (rounded appearance) and faster degeneration indicate that excess wild-type p53 protein may cause apoptotic change, a finding similar to that of a previous report (Clarke et al. 1993). The finding that transfecting both wild-type and mutated 280 p53 genes with a 1-to-1 ratio into the Saos-2 cells caused the wt-p53 and mt 280-p53 protein complex to appear in their nuclei (by immunohistochemistry) suggests that the Saos-2 cells do not contain the specific cytoplasmic factor(s) to hold the p53 protein in their cytoplasm, unlike the NPC cells.
The mechanism(s) of cytoplasmic accumulation of p53 protein in NPC cells is not yet well defined (Bosari et al. 1995; Stenmark Askmalm et al. 1994; Moll et al. 1992, 1995). However, a defect of nuclear localization in p53 protein or an aberrant nuclear trans-location pathway is unlikely, because under starvation conditions the mutant p53 protein can still be transported into NPC nuclei (Figure 1B). We suspect that certain unidentified cellular protein factors may play a role in binding and anchoring the mutated p53 protein in the cytoplasm, as mentioned above. Further investigation is needed to identify these cellular factor(s).
Although cancer cells treated with cisplatin may induce nuclear accumulation of wt p53 (Fritsche et al. 1993), in our NPC cells, after treatment with cisplatin, the 280 mt p53 protein is still localized in the cytoplasm. When cisplatin was used to treat the wt p53 and 280 mt p53 transfectants, the survival rate in each condition was similar, indicating that 280 mt p53 protein does not cause the NPC tumor cells to become resistant to therapy, unlike the mutant p53 in other tumor cell types (Lowe et al. 1994).
Footnotes
Acknowledgements
Supported in part by a research grant (NSC85-2331-B-002-188-M06) from the National Science Council (CTL), by one from Academia Sinica, Taipei (CTL), and another from National Taiwan University Hospital (INTPRO-08) (JKH), Taipei, Taiwan, R. O. C.
We are grateful to Drs Y.S. Lin and T. Tan of the Institute of Biomedical Sciences, Academia Sinica, Taipei, and to Dr J.Y. Shew of Department of Biochemistry, College of Medicine, National Taiwan University, for helpful criticism. We also thank Ms H.M. Hwang for technical assistance.