
Abstract
We have developed enhanced immunohistochemical protocols for detecting autonomic nerve fibers and splenocyte-associated proteins in rat spleen. This includes norepinephrine-synthesizing enzymes (dopamine-βeta hydroxylase (DBH) and tyrosine hydroxylase (TH)), neuropeptide Y (NPY), tumor necrosis factor -α (TNF-α), interferon-γ (IFN-γ), c-fos protein, inducible nitric oxide synthase (iNOS), and the macrophage cell marker ED1. Animals were divided into sham-operated and splenic nerve-sectioned groups for detection of DBH, TH, and NPY. For immunodetection of TNF-α, iNOS, IFN-γ and c-fos, animals were injected IV with saline or 100 μg of lipopolysaccharide (LPS) and were sacrificed at various time intervals post injection. Rats were perfused with 4% paraformaldehyde, spleens removed and cryoprotected, and 50-μm floating sections were cut on a freezing microtome. Immunodetection was performed with various detection systems and substrate/chromogen solutions, and in some cases using pretreatment with proteinase K (PK) for antigen unmasking. PK pretreatment increased immunostaining for DBH, TH, NPY, IFN-γ, iNOS, and ED1, and the improvement was concentration-dependent. Using NPY immunostaining to index the signal-to-noise ratio for various substrates and detection systems, we found that an alkaline phosphatase detection system with NBT/BCIP as a substrate was the best procedure for light microscopy, whereas the CY3-labeled secondary antibody technique proved optimal for fluorescent microscopy. Surgical transection of the splenic nerve eliminated all nerve fiber staining for DBH, TH, and NPY. TNF-α, IFN-γ, c-fos, and iNOS proteins were observed in the spleen in a time-dependent manner after LPS stimulation. Fluorescent double labeling, visualized with fluorescent confocal scanning laser microscopy, revealed many NPY fibers distributed among the ED1-labeled macrophages. These results demonstrate that immunohistochemistry can be used to index the activational effects of an immune challenge on splenocytes in situ and verifies that splenic immune cells are innervated by the sympathetic nervous system.
Keywords
Neuroanatomic and neuroendocrine studies have demonstrated a role for the nervous system in regulating immune function via the hypothalamic-pituitary-adrenal (HPA) axis and the autonomic nervous system. The sympathetic arm of the autonomic nervous system may play a major role in regulating immune function via direct sympathetic innervation of all immune organs (Trudrung et al. 1994; Nance and Burns 1989; Felten et al. 1987a,b). Typically the sympathetic nervous system exerts inhibitory control of immune function (Hu and Moller 1994; Dureus et al. 1993; Monastra and Secchi 1993; Wan et al. 1993b; Hu et al. 1991; Kouassi et al. 1988; Besedovsky et al. 1979). However, some immune responses have been shown to be potentiated by sympathetic activation (Zalcman et al. 1994).
The spleen is a model organ to study neural-immune interactions because of its well-described innervation (Trudrung et al. 1994; Nance and Burns 1989) and the ability to eliminate nerve fibers to the spleen by chemical or surgical sympathectomy (Zalcman et al. 1994; Vriend et al. 1993; Wan et al. 1993b; Romano et al. 1991; Nance and Burns 1989; Felten et al. 1987a,b; Besedovsky et al. 1979). Analysis of the effects of neural transmitters on splenic immune function indicate a functional role for norepinephrine (NE) and neuropeptide Y (NPY) (Hu and Moller 1994; Madden et al. 1994; Zalcman et al. 1994; Dureus et al. 1993; Fukushima et al. 1993; Monastra and Secchi 1993; Wan et al. 1993b; Hu et al. 1991; Spengler et al. 1990; Kouassi et al. 1988; Sanders and Munson 1985). Of particular interest are the effects of NE agonists and antagonists on the in vitro production of macrophage-associated cytokines, such as tumor necrosis factor-α (TNF-α) (Monastra and Secchi 1993; Spengler et al. 1990; Introna et al. 1986). Although these in vitro studies provide information on possible cellular interactions between sympathetic neural transmitters and the immune system, they may not accurately reflect the events that occur in vivo. Therefore, we have developed enhanced immunohistochemical protocols that enable us to examine the role of the autonomic nervous system in regulating splenic immune function by in situ localization of immune-related molecules and autonomic nerve fibers.
Dilutions of primary antibodies
Materials and Methods
Chemicals
Lipopolysaccharide (LPS; E. coli serotype 055:B5), diaminobenzidine, glucose oxidase, nitroblue tetrazolium (NBT), levamisole, d-glucose, Fast Red, naphthol AS-MX phosphate, sodium nitroprusside, proteinase K (PK), glycine, BSA, and Triton X-100 were purchased from Sigma (St Louis, MO). Paraformaldehyde, sodium nitrite, and glycerol were purchased from BDH (Toronto, Ontario, Canada), 5-bromo-4-chloro-3-indolyl-phosphate-p-toluidine salt (BCIP), ammonium chloride, N,N-dimethylformamide, sodium azide, methanol, and gelatin were purchased from Fischer (Fair Lawn, NJ). The Enzyme-Labeled-Fluorescence (ELF) kit was purchased from Molecular Probes (Eugene, OR). Normal goat serum was purchased from Cappel (Scarborough, Ontario, Canada) and RedPhos was purchased from Research Organics (Cleveland, OH).
Antibodies
Rabbit anti-c-fos was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-dopamine-β hydroxylase (DBH) and anti-tyrosine hydroxylase (TH) were purchased from Eugene Tech (Ridgefield Park, NJ). Rabbit anti-NPY was purchased from Incstar (Stillwater, MN). Rabbit anti-inducible nitric oxide synthase (iNOS) antibodies were a gift from Dr. H. Ohshima (International Agency for Research on Cancer; Lyon, France) or purchased from Transduction Labs (Lexington, KY). Rabbit anti-mouse TNF-α was purchased from Genzyme (Cambridge, MA). Rabbit anti-rat interferon-γ (IFN-γ) was purchased from Biosource (Camarillo, CA). Rabbit IgGs were purchased from Sigma, CY3-labeled goat anti-rabbit and FITC-labeled goat anti-mouse were purchased from Jackson Immunologicals (West Grove, PA), mouse monoclonal anti-ED1 was purchased from Serotec (Toronto, Ontario, Canada), mouse IgGs were purchased from Rockland Labs (Gilbertsville, PA), and unconjugated and alkaline phosphatase (AP)-conjugated goat anti-rabbit and rabbit peroxidase-anti-peroxidase (PAP) were purchased from Cappel (Scarborough, Ontario, Canada). Optimal dilutions of primary and secondary antibodies were determined empirically in preliminary experiments. Listed in Tables 1 and 2 are the optimal dilutions of primary and secondary antibodies with the AP detection system using NBT/BCIP as a chromogen/substrate combination. References documenting the specificity of these antibodies also appear in Table 1.
Dilutions of secondary antibodies
Animals
For immunodetection of DBH, TH, and NPY fibers in the spleen, adult male (250–400 g) Sprague-Dawley rats (Charles River; Dorval, Quebec, Canada) were anesthetized (60 mg/kg sodium pentobarbital) and divided into two groups, sham surgeries and splenic nerve sections (described by Nance and Burns 1989) and allowed to recover for 7–10 days before tissue processing (six animals per group). For studies involving the immunodetection of TNF-α, c-fos, IFN-γ, and iNOS, animals were injected with saline or 100 μg of LPS via tail vein (at least three animals per time point) and were sacrificed at various intervals after injection. All procedures were approved by the animal ethics committee at the University of Manitoba and the CCAC.
Tissue Processing
Animals were sacrificed by an overdose of pentobarbital and then transcardially perfused with 100 ml of 1% sodium nitrite in phosphate buffer (PB), followed by 300 ml of 4% buffered paraformaldehyde (pH 7.3). Spleens were removed, postfixed for 2 hr, and cryoprotected in 30% sucrose. Serial 50-μm sections of spleen were cut on a freezing microtome and transferred to a 24-well culture plate containing 0.01 M PBS. Subsequent processing of the sections depended on the enzymatic or fluorescent detection system being tested.
PAP Detection
Sections were rinsed three times or were pretreated with 0–5 μg/ml of proteinase K (PK 0–5 μg/ml) in PK buffer (0.1 M Tris/50 mM EDTA, pH 8.3, at 25°C) for 30 min at 37C with agitation before the rinse steps (the first rinse after PK included 2 mg/ml glycine). Next, sections were either placed in primary antibody or were pretreated with 3% H2O2 and 0.1% azide (Li et al. 1987) at room temperature (RT) for 30 min to reduce endogenous peroxidase activity before being placed in primary antibody. Primary antibodies were diluted in 0.01 M PBS containing 2% bovine serum albumin (BSA), 1% normal goat serum (NGS), and 1% Triton X-100 (TTX). Trays were enclosed in humidified bags on a rocker table and incubated overnight at RT. The next day all sections were rinsed and placed in PBS/1% TTX/1% NGS for 90 min containing 1:150 dilution of unconjugated goat anti-rabbit antibody. Sections with and without previous pre-treatment to reduce endogenous peroxidase activity were incubated for 30 min in 3% H2O2/ 5% methanol in PBS (Romano et al. 1991) or 1% sodium nitroprusside/ 0.074% HCl/100% methanol (Straus 1971) and rinsed. The sections were placed in 1:300 dilution of PAP in PBS/1% TTX/1% NGS for 90 min. Sections were rinsed and developed in 500 μl of 0.01 M PB/well containing 0.05% diaminobenzidine, 0.04% ammonium chloride, and 0.2% d-glucose. After addition of 50 μl of 0.006% glucose oxidase solution to each well, the sections were developed for 30–45 min and then rinsed in PBS, floated onto slides, dried overnight, and coverslipped in glycerol gel (50% glycerol/ 7.5% gelatin/0.1% azide in 0.1 M PB).
Alkaline Phosphatase Detection
Sections were rinsed and placed directly in primary antibody or first digested with PK as described above. After incubation in primary antibody, sections were rinsed and placed into a 1:750–1:1000 dilution of AP-conjugated goat anti-rabbit [whole antibody or F(ab)2 fragments] for 2 hr, rinsed, and then developed with one of the following substrates: NBT/BCIP, NBT/Red-phos, Fast Red, or ELF (Larison et al. 1995). For NBT/BCIP and NBT/RedPhos detection, 0.4 mM NBT, 0.4 mM BCIP (or RedPhos), and 3 mM levamisole were added to 50 mM MgCl2/100 mM Tris/100 mM NaCl, pH 9.3. Fast Red was developed by dissolving 20 mg of Fast Red salt in 20 ml of 100 mM Tris/3 mM levamisole (pH 8.2). To this solution, 4 mg ASMX-phosphate dissolved in 400 μl of dimethylformamide was added and stirred for 30 sec without filtering. ELF development was according to the manufacturer's instructions, except that the development solution was not filtered and sections were developed for 5–10 min. After development, sections were rinsed, floated onto slides, dried, and coverslipped as above.
CY3 Detection
Sections were rinsed and placed in primary antibody or pretreated with proteinase K as described above. After the primary incubation, sections were rinsed and incubated for 3-4 hr in a 1:1000 dilution of CY3-conjugated goat anti-rabbit antibody. Sections were rinsed, floated onto slides, air-dried, and coverslipped as above.
Fluorescent Double Labeling
Sections were incubated overnight with rabbit anti-NPY mixed with mouse anti-ED1 in 1% NGS, 2% BSA, and 1% TTX. Sections were then rinsed with PBS and incubated for 3-4 hr in goat anti-rabbit labeled with CY3 and goat anti-mouse labeled with FITC. Sections were rinsed in PBS and mounted as described above. Sections were then visualized with a Leitz epifluorescent microscope and with a Molecular Dynamics confocal scanning laser microscope equipped with an argon laser and dual detectors. The images were generated from unfiltered raw optical sections that were rendered as maximal intensity projections with ImageSpace software (Molecular Dynamics; Sunnyvale CA).
Controls
Control staining procedures included exclusion of primary and/or secondary antibodies and nonspecific rabbit or mouse immunoglobulins substituted for primary antibodies. The same controls were applied for double-labeling experiments, except that rabbit IgG and antibody to NPY were incubated with goat anti-mouse-conjugated FITC and mouse IgG and anti-ED1 were incubated with CY3-labeled goat anti-rabbit to check for crossreactivity.
Results
Immunohistochemistry for DBH with the PAP and AP Techniques
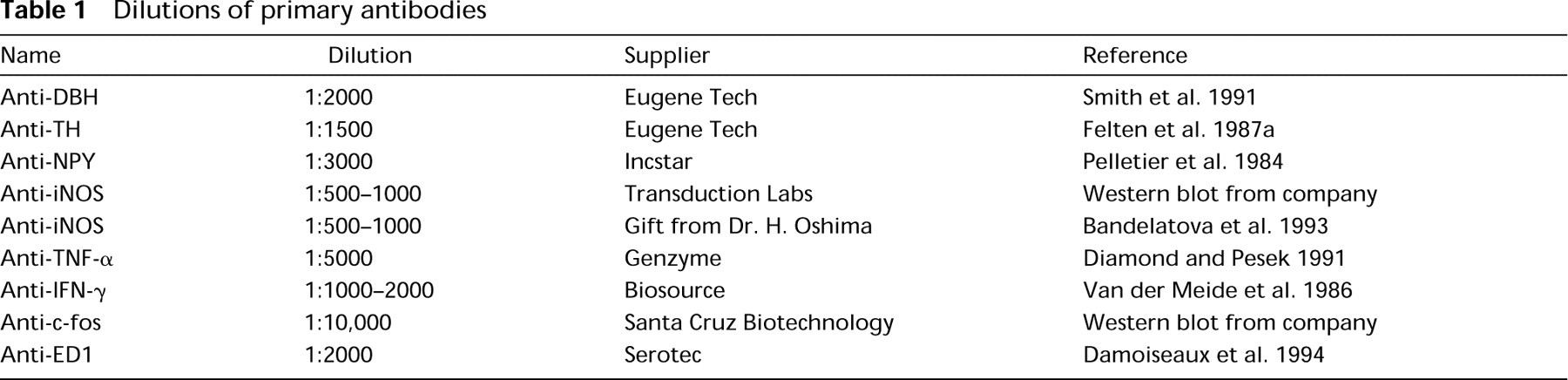
The focus of this study was to determine the location of nerve fiber- and splenocyte-associated proteins in rat spleen. Initially the PAP technique was used for immunolocalization of DBH and other molecules, but it was unsuitable because of the low signal-to-noise ratio. We observed that the spleen has two sources of background with the PAP technique: a diffuse pseudo-peroxidase activity, probably originating from red blood cells and granulocytes, and clusters of very strongly positive cells located in the red pulp and marginal zones. Although the diffuse background staining did not significantly impair the analysis of results, the strongly staining clusters of cells posed a major problem for analyzing cell-associated molecules. In an attempt to improve immunodetection with the PAP technique, we investigated various methods for reducing endogenous peroxidase activity (EPA), including peroxide (H2O2)/azide, H2O2/methanol, methanol/HCl/sodium nitroprusside, and a combination of H2O2/azide and H2O2/methanol. We found that all four methods reduced EPA to a similar extent, but the combination of H2O2/azide with H2O2/methanol consistently produced the best results in terms of signal-to-noise ratios (unpublished observations). The H2O2/methanol method was marginally better than H2O2/azide at reducing EPA, but H2O2/azide was better for preservation of tissue morphology. The third method, nitroprusside/methanol/HCl, was very effective at eliminating EPA, but it also decreased positive signal and strongly affected tissue morphology. Diluting the secondary antibody (goat anti-rabbit) or the PAP complex helped to reduce background staining in some spleen sections but often reduced positive staining as well. Some spleen sections continued to show clusters of cells with strong EPA, even after various bleaching methods, omission of antibodies, and PK treatment. Together, these observations indicated that tissue thickness, nonspecific binding of antibodies to Fc receptors and other antigens, and EPA were all likely contributors to the background staining with the PAP technique. In contrast to the PAP technique, the AP technique (Figure 1) demonstrated a high signal-to-noise ratio with minimal interference from endogenous AP activity. In side-by-side comparisons, the AP detection system was always qualitatively and esthetically superior to the PAP technique.
Photomicrographs showing endogenous peroxidase and endogenous alkaline phosphatase activity and the effect of proteinase K pretreatment on DBH immunostaining in the spleen with the PAP procedure. (
Antigen Unmasking with Proteinase K
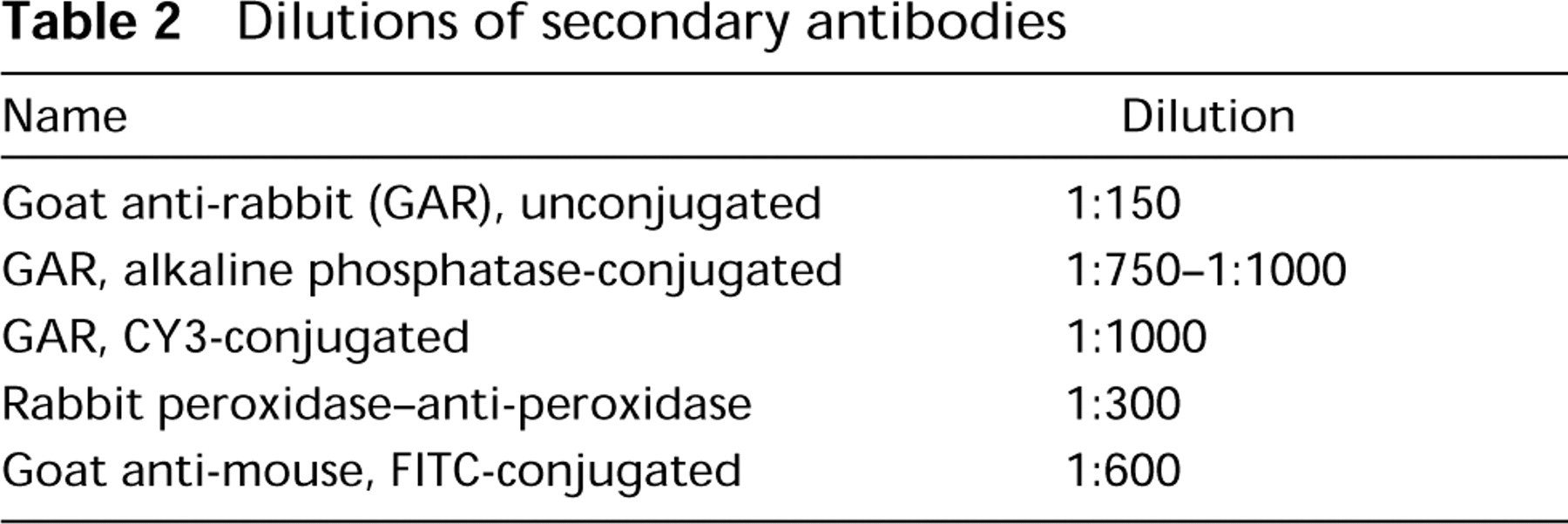
The staining for DBH with both the PAP (Figure 1C) and the AP (Figure 2A) detection system was weak in undigested tissue sections. However, a dramatic and concentration-dependent improvement was observed for DBH staining with both detection systems if the spleen sections were pretreated with PK (Figures 1D and 2B-2D). Similar observations were made with TH and NPY (not shown).
Photomicrographs showing the effect of different proteinase K (PK) concentrations on DBH immunodetection, as visualized by an alkaline phosphatase detection procedure with NBT/BCIP as substrate. (
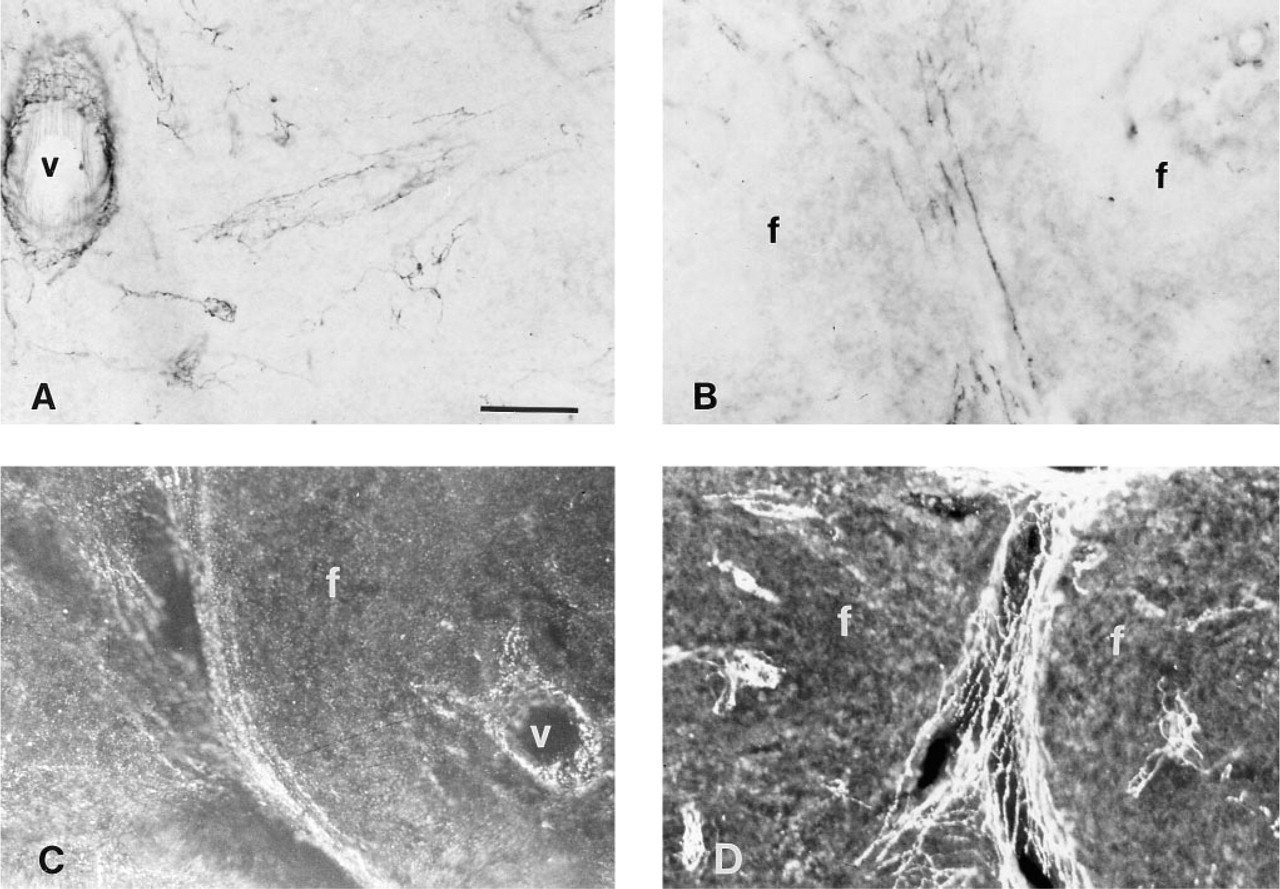
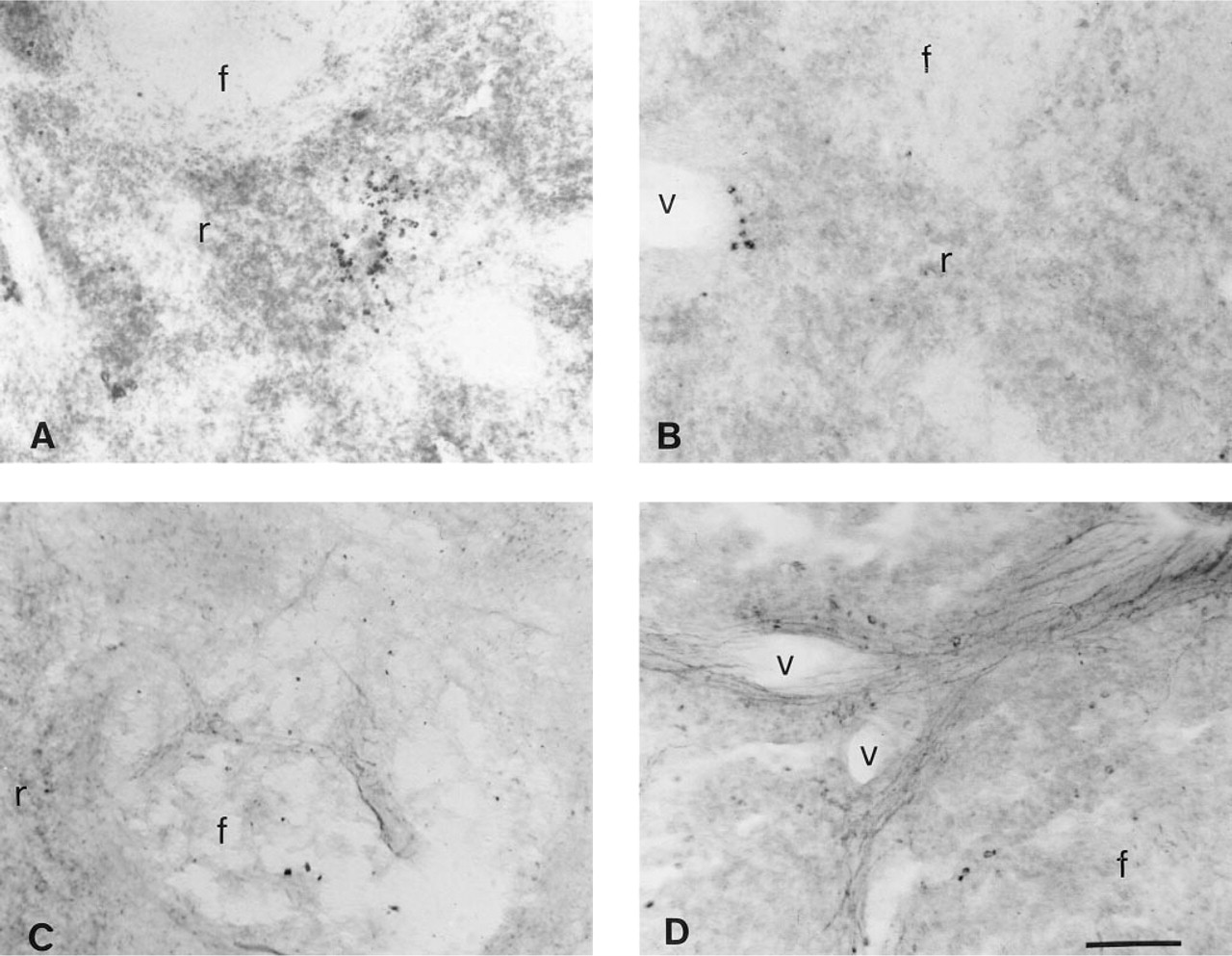
Photomicrographs showing immunostaining of PK-treated spleen sections for NPY-positive nerve fibers using different substrates and detection systems other than an alkaline phosphatase detection system with NBT/BCIP. (
Comparison of Different Substrates for NPY Detection
To determine the optimal detection system and substrate, we compared NPY-positive immunodetection using AP substrates other than NBT/BCIP, as well as other detection systems (Figure 3). The substrates included NBT/RedPhos, Fast Red, and ELF, and the detection systems include the PAP technique and a fluorescent-conjugated antibody. Among the AP substrates, we found that NBT/BCIP was superior. Although the RedPhos worked well for NPY immunohistochemistry, it did not develop as quickly or stain as intensely as did NBT/BCIP. The Fast Red substrate could be visualized by light and fluorescence microscopy, but it produced a weak signal for both methods of visualization (not shown). Although detection of NPY with the ELF kit proved to be the better of the two fluorescent AP substrates, a CY3-labeled secondary antibody gave optimal results for NPY immunostaining. CY3 provided a high contrast with intense red nerve fibers against a dark background, and was highly resistant to photobleaching.
Effect of Splenic Nerve Sectioning on Immunostaining for Nerve Fibers
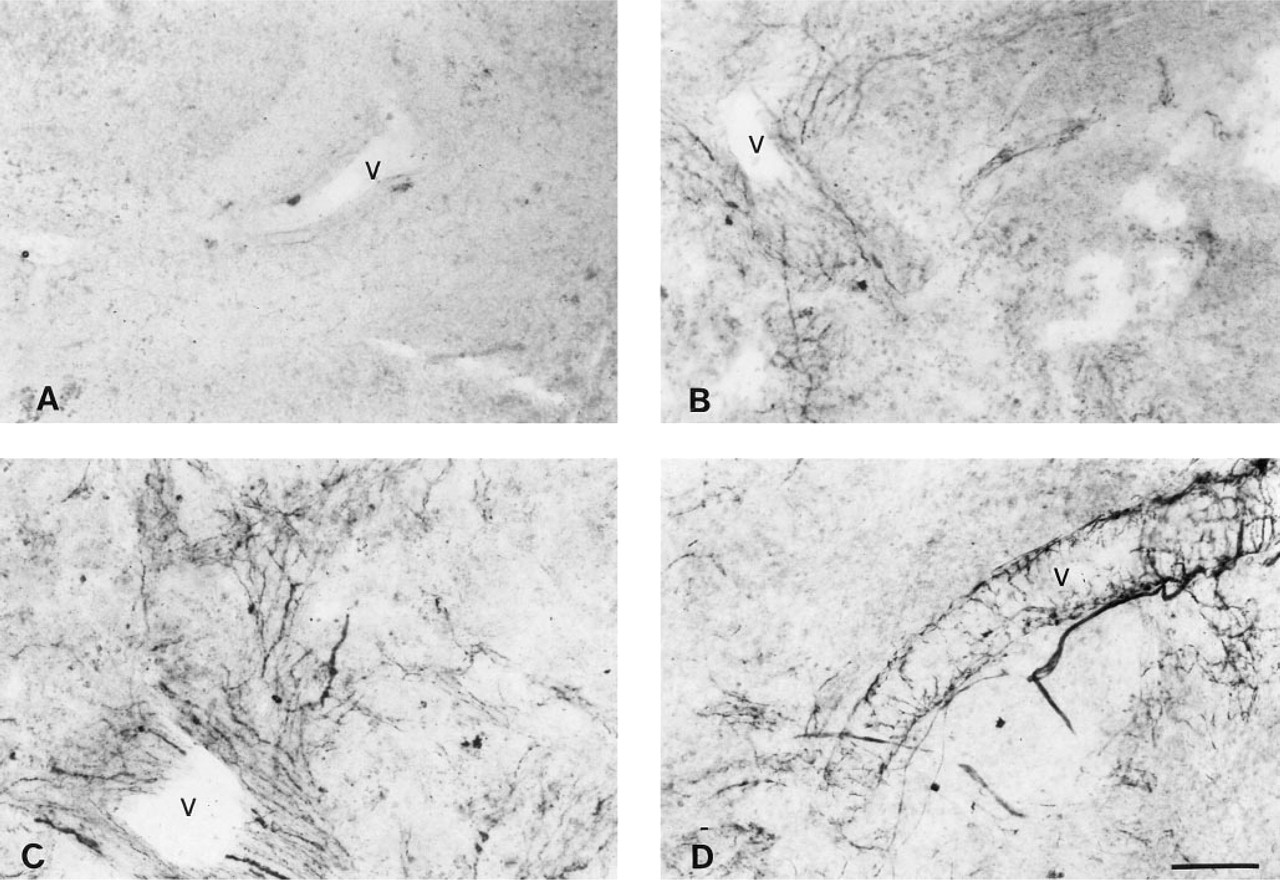
Using the AP system with NBT/BCIP, we demonstrated intense immunostaining for DBH, TH, and NPY in the spleens of sham-operated animals and a complete absence of staining for these molecules in nerve-sectioned animals (Figure 4).
Detection of TNF-α, IFN-γ, c-fos, and iNOS After LPS Injection
The results demonstrated that only a few TNF-α-positive cells and no c-fos or IFN-γ-positive cells were detectable in saline-treated rats. However, after LPS injections, many positive cells were observed for TNF-α, IFN-γ and c-fos, distributed in a parafollicular pattern (Figure 5). We observed a major increase in TNF-α-positive cells from 30 min post LPS until 2 hr post LPS. C-fos-positive cells were first detected at 1 hr post LPS and maintained this intensity until 2 hr post LPS. At 4 hr post LPS there were no TNF-α-positive cells and only a few c-fos-positive cells. However, many IFN-γ-positive cells were present at 4 hr post LPS. A few iNOS-positive cells were present in saline-injected animals, but we observed a dramatic and time-dependent increase in iNOS staining in the spleen starting at 4 hr post LPS with maximal staining being observed at 6 hr post LPS (Figure 6). Antigen unmasking with PK improved immunostaining for iNOS, IFN-γ (not shown), and the macrophage cell marker ED1 (not shown) in the spleen.
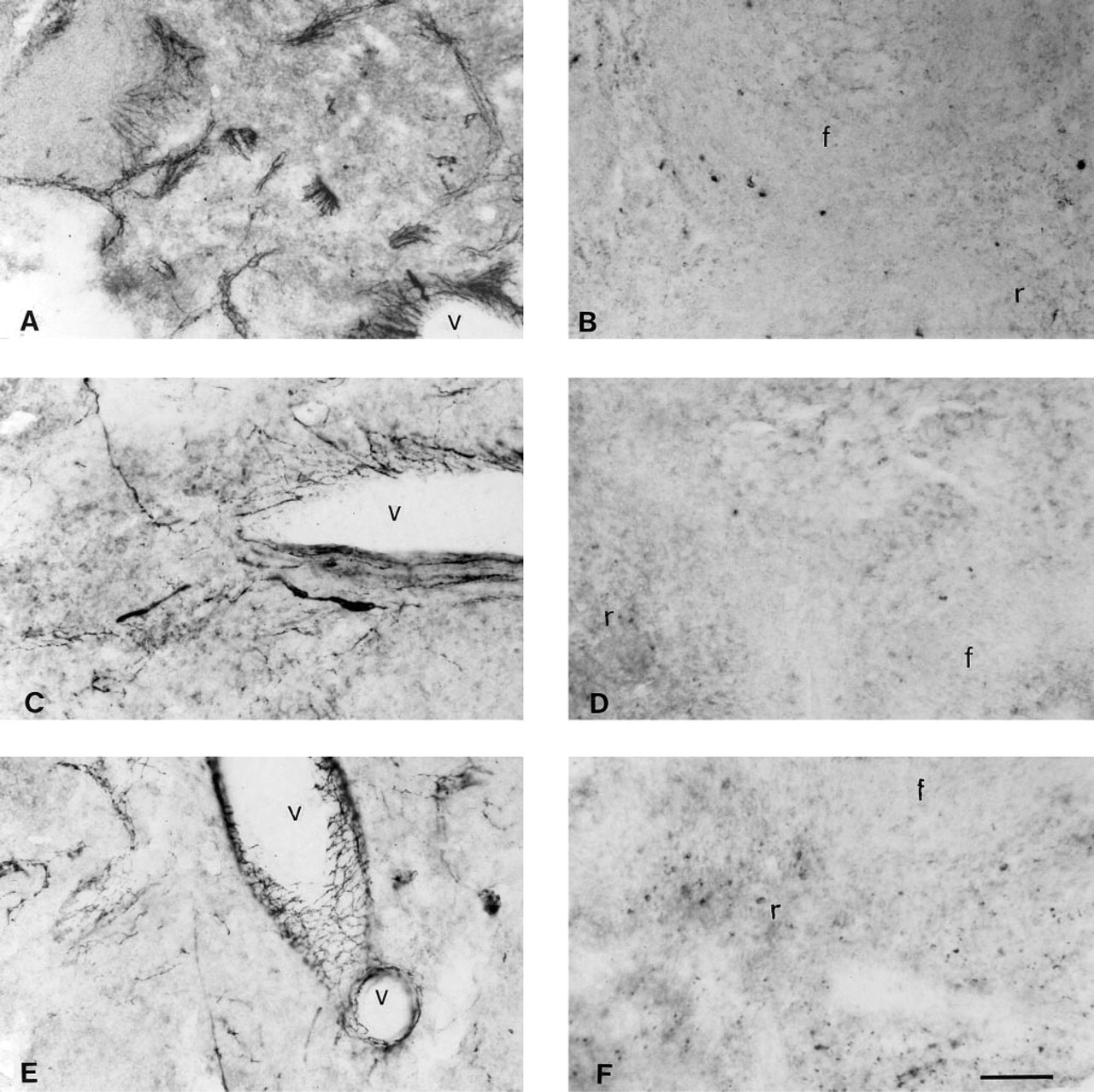
Photomicrographs showing DBH, TH, and NPY immunostaining in spleens of control and splenic nerve-sectioned animals. (
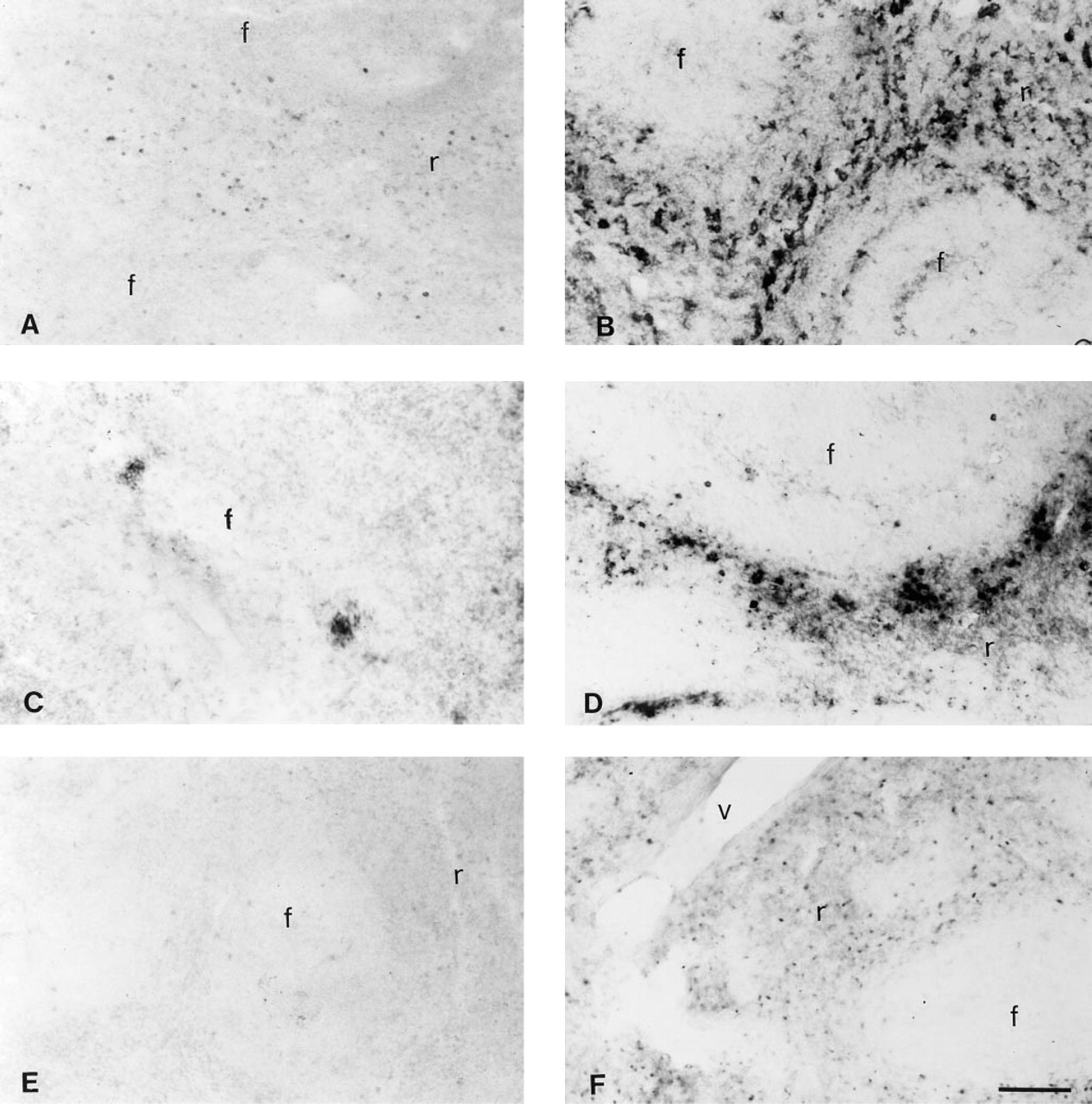
Photomicrographs showing TNF-α, IFN-γ and c-fos immunostaining in spleens of saline and LPS-treated animals. (
Co-localization of NPY-positive Fibers with ED1-positive Cells
Using confocal microscopy, we demonstrated that macrophages (ED1-positive) are located in the same tissue compartment in the spleen as the sympathetic nerve fibers (NPY-positive) (Figure 7). This verifies previous anatomic and immunological studies that suggest a role for the sympathetic regulation of immune function.
Discussion
Although the PAP detection protocol provided excellent sensitivity for DBH (Figure 1D) and NPY (Figure 3A) immunodetection, the unpredictability of cellular background and the extra steps needed to remove this background suggested that it was not the optimal detection system for cell-associated antigens in the spleen. Both the AP detection system with NBT/BCIP as a substrate and the CY3-conjugated antibody gave the best signal to noise ratio for detection of autonomic nerve fibers and immune-related proteins in the spleen. We also demonstrated that PK digestion dramatically improves nerve fiber staining for DBH, TH, and NPY and cellular staining for iNOS (Figure 6), IFN-γ, and ED1 (not shown). The optimal dose of PK to use for immunohistochemistry was variable among the different primary antibodies and even between groups of spleens perfused at different times, but the effective concentrations were 0–5 μg/ml, with the limiting step being the integrity of the tissue.
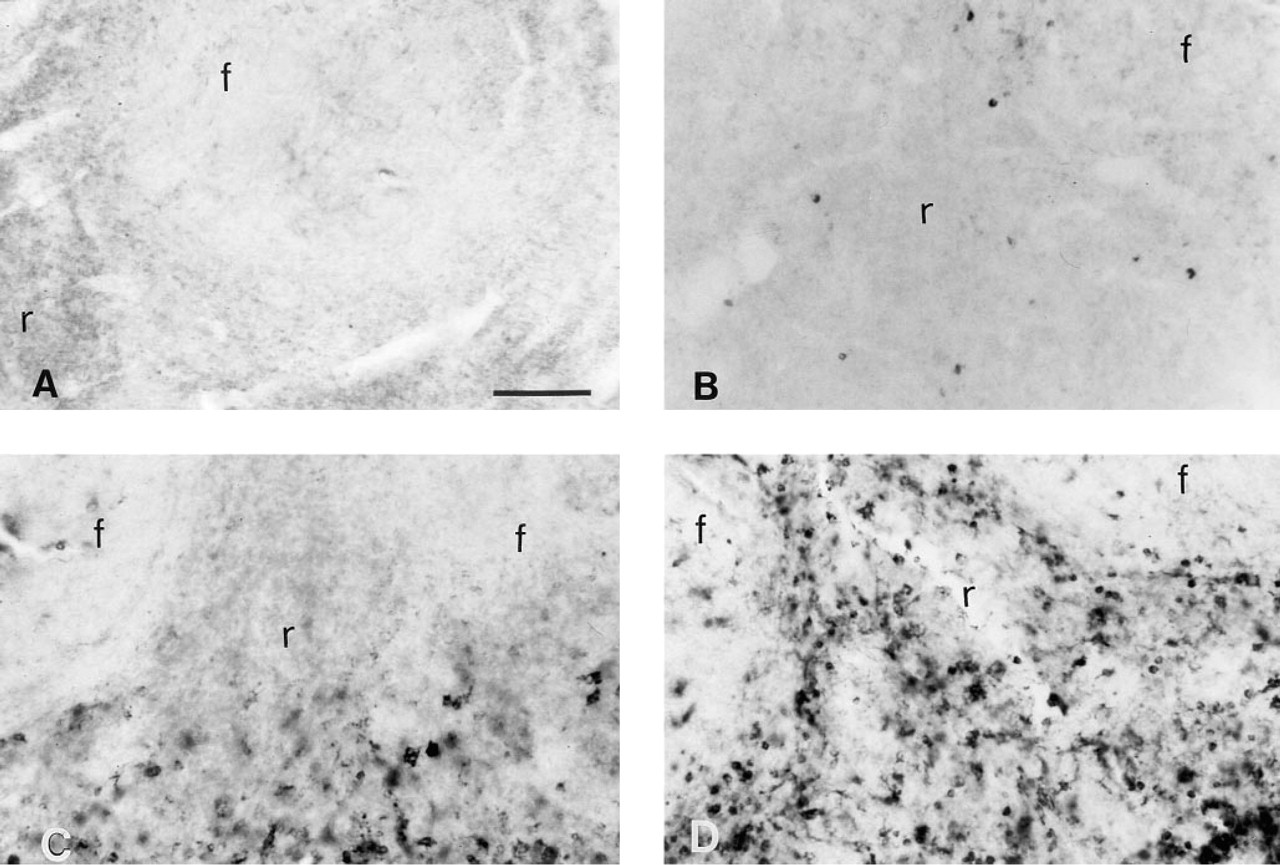
Photomicrographs showing iNOS immunostaining in spleens of saline- or LPS-treated rats with and without PK pretreatment. (
The use of proteolytic enzymes such as PK for antigen unmasking in immunohistochemistry has been described (Polak and Van Noorden 1982). However, the use of PK for improvement in nerve fiber immunodetection in the spleen is novel. Although it is unknown exactly why PK treatment enhances the immunostaining of some molecules, it is likely that several factors play important roles, including size and subcellular location of the molecule, type and amount of fixation, and the epitope recognized by the antibody.
Using optimized immunodetection techniques, we demonstrated that transection of the splenic nerve eliminated catecholamine-containing fibers in the spleen. This verifies previous data (Vriend et al. 1993) that showed a 98% depletion of NE levels in surgically denervated spleens, as measured by HPLC. Therefore, surgical sympathectomy is an effective method for removing the sympathetic control of the spleen and, relative to chemical sympathectomy, provides much greater anatomic specificity.
We also localized cytokines such as TNF-α (Diamond and Pesek 1991; Brown and Fishman 1990; Hofsli et al., 1989; Chensue et al. 1988) and IFN-γ (Heinzel et al. 1994; Heremans et al. 1994), transcription factors such as c-fos (Wan et al. 1993a, 1994; Hamilton et al. 1989; Collart et al. 1987; Introna et al. 1986) and enzymes such as iNOS (Sato et al. 1995; Buttery et al. 1994; Cook et al. 1994; Bandaletova et al. 1993; Marletta 1993) in the spleen after injection of LPS (Hewett and Roth 1993). Only a few cells expressed the proteins for TNF-α, IFN-γ, c-fos and iNOS in rats injected with saline. However, after LPS stimulation, many cells were immunopositive for these proteins in the marginal zone and red pulp, suggesting that they are probably macrophagic in origin and, in the case of IFN-γ, possibly NK- or T-cells (Heinzel et al. 1994; Heremans et al. 1994). We also demonstrated that ED1-positive cells can be co-localized with NPY-positive nerve fibers with immunofluorescence. Studies using confocal scanning laser microscopy demonstrated functional co-localization of sympathetic nerve fibers with immune effector cells in the spleen, confirming the anatomic co-localization reported by Felten et al. (1987a,b).
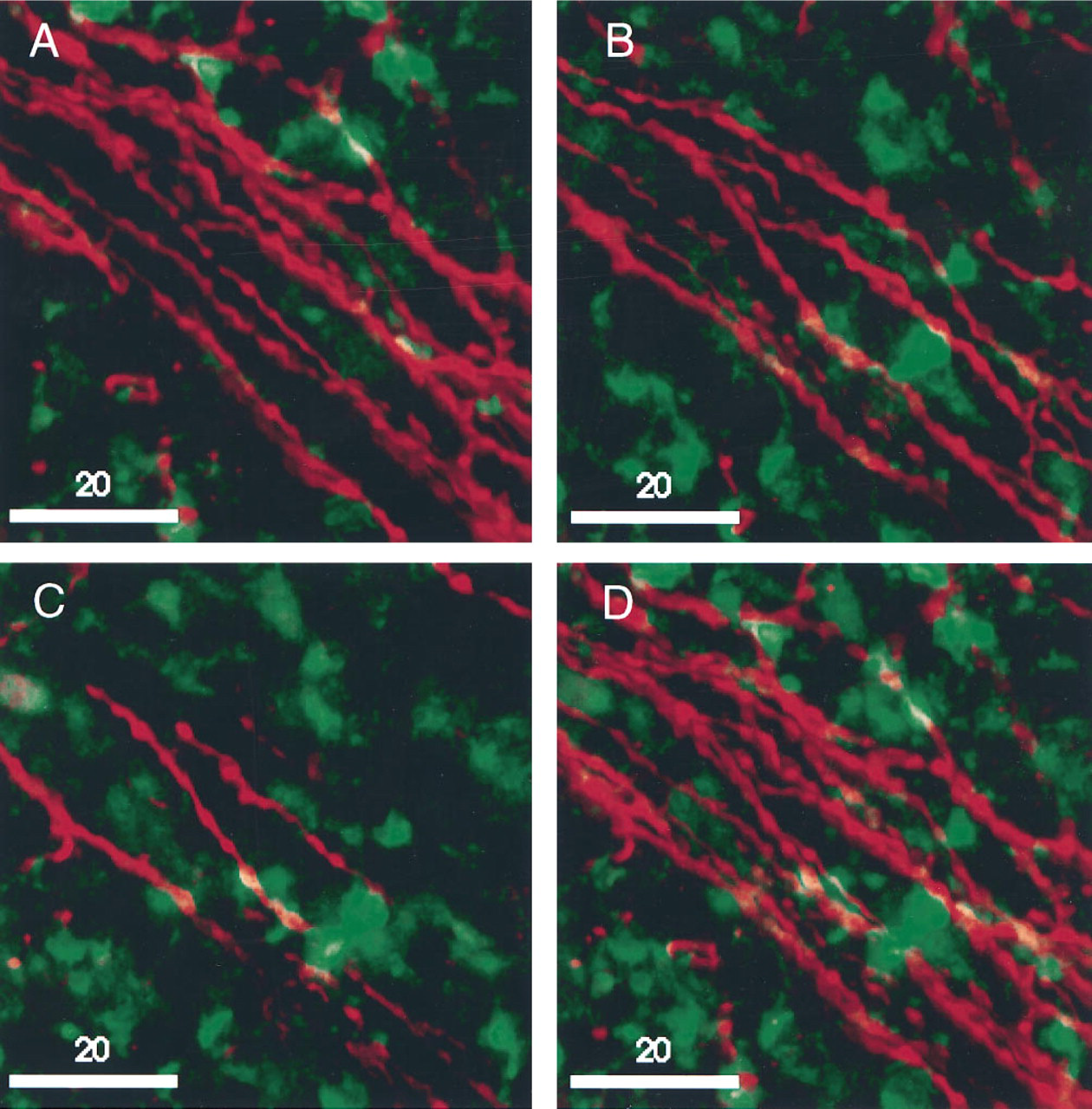
Digital prints of dual confocal microscope images of ED-1-positive macrophage (FITC) and NPY-positive nerve fibers (CY3) in rat spleen, visualized with a Molecular Dynamics confocal scanning laser microscope equipped with an argon laser and dual detectors.
In conclusion, this article describes the ability to verify splenic nerve sections, to detect a variety of LPS-inducible proteins and to localize these proteins, to specific cell types by immunohistochemical techniques. More importantly, this methodology may provide a valuable dependent measure of in situ immune function that can be utilized to assess the influence of the sympathetic nervous system on immune function.
Footnotes
Acknowledgements
Supported by grant no. MH4 3778-04A2 from the National Institutes of Health, Bethesda, MD.
We thank Dr A. Jansen, Dr B. MacNeil, V. Sanders, S. Pylypas, and E. Stern for technical assistance, Dr H. Ohshima for his generous gift of the rabbit anti-iNOS antibody, and Dr C. Braekevelt for his gift of the anti-ED1 antibody.