
Abstract
Transforming growth factor-β (TGF-β) depresses mucosal inflammation and upregulates extracellular matrix (ECM) deposition. We analyzed TGF-β receptors RI and RII as well as ECM components using the CD4+ T-cell-transplanted SCID mouse model of colitis. The principal change in colitis was an increased proportion of TGF-β RII+ mucosal mesenchymal cells, predominantly α-smooth muscle actin (SMA)+ myofibroblasts, co-expressing vimentin and basement membrane proteins, but not type I collagen. TGF-β RII+ SMA− fibroblasts producing type I collagen were also increased, particularly in areas of infiltration and in ulcers. Type IV collagen and laminin were distributed throughout the gut lamina propria in disease but were restricted to the basement membrane in controls. In areas of severe epithelial damage, type IV collagen was lost and increased type I collagen was observed. To examine ECM production by these cells, mucosal mesenchymal cells were isolated. Cultured cells exhibited a similar phenotype and matrix profile to those of in vivo cells. The data suggested that there were at least two populations of mesenchymal cells responsible for ECM synthesis in the mucosa and that ligation of TGF-β receptors on these cells resulted in the disordered and increased ECM production observed in colitic mucosa.
T
TGF-β activity is tightly regulated both during proteolytic generation of the biologically active cytokine (reviewed Keski–Oja et al. 1995) and also at the level of the cellular response to active TGF-β (reviewed Kolodziejczyk and Hall 1996), through selective receptor expression, control of receptor density, and subsequent intracellular signaling. After activation via proteolytic cleavage, TGF-β operates via two major signaling receptors, type I and type II (RI and RII). There are also four other known receptors. Betaglycan (formerly RIII) (Lopezcasillas et al. 1993) and endoglin (CD105) (St-Jacques et al. 1994) are non-signaling receptors that increase the affinity of RII for TGF-β, RIV, which has a limited distribution and unknown function, and RV, which can signal together with RI but also in the absence of both RI and RII (Liu et al. 1997). TGF-β binds to RII, which then phosphorylates RI and initiates a downstream intracellular signaling cascade via the smad proteins (reviewed in Roberts 1999), but smad-independent routes are also used (Choi et al. 1999). Changes in receptor expression may be a factor in disease pathologies. For example, decreased RII has been noted in carcinomas (Jakowlew et al. 1998), leading to uncontrolled proliferation, and inflammatory stimuli decrease TGF-β receptor expression in monocytes (Brandes et al. 1991). Increased RII has been observed in fibrosis and tissue repair (Schmid et al. 1998; Menke et al. 1999), possibly contributing to increased ECM deposition.
There are contradictory reports on intestinal TGF-β receptor expression. In human gut, RI and RII have been reported to be expressed by colon surface epithelial cells (Eskinazi et al. 1998; Planchon et al. 1999), but these reports did not concur on lamina propria (LP) cell expression. In the rat, most expression was observed, by Western blotting of fractionated cells, in the non-epithelial compartment and, by immunohistology, in the lamina propria and muscle (Winesett et al. 1996). In developing rat gut, RI and RII are expressed on epithelial cells and muscle but are expressed at low level in the gut LP, whereas RIII is expressed on several types of immune cells in the LP (Zhang et al. 1999). Endoglin is expressed on many stromal cells in mouse gut, particularly endothelium (St-Jacques et al. 1994). In the normal mammalian gut, epithelial and muscle cells express high levels of signaling receptors, whereas stromal cells express high levels of molecules that increase the affinity of RII for its ligand. LP mononuclear cells presumably express signaling receptors in vitro, because TGF-β has been shown to rescue mitogen-stimulated LP CD4+ T-cells from apoptosis (Plunkett 2000), and phosphorylation of smad 3 in isolated colon LP mononuclear cells has been demonstrated (Monteleone et al. 2001).
The human inflammatory bowel diseases (IBD), Crohn's disease and ulcerative colitis, are chronic diseases of unknown etiology but are probably caused by a loss of immunological tolerance, mediated by T-cells, to the normal indigenous flora (Fiocchi 1998). Animal models of IBD support this concept (Blumberg et al. 1999), and TGF-β has been shown to be a key molecule in mediating mucosal tolerance (Chen et al. 1995). An inappropriate distribution or amount, either of active TGF-β or of TGF-β receptors, could account for the accumulation of T-cells and ECM changes observed in Crohn's disease. The distribution of TGF-β receptors in severe Crohn's disease is increased in LP lymphocytes, epithelial cells, and fibroblasts (Di Mola et al. 1999), but this study detected very little expression of any of these components in control tissue, in contrast to several other studies (Eskinazi et al. 1998; Planchon et al. 1999).
The transfer of CD4+ T-cells into severe combined immunodeficient (SCID) mice results in a Th1-mediated colitis with similarities to Crohn's disease (transmural involvement, granuloma formation, mucosal hyperplasia, and T-cell accumulation in the mucosa) (Powrie et al. 1993; Rudolphi et al. 1994). In previous work using this model (Whiting et al. 2001), we explored the hypothesis that a defect in the mucosal TGF-β system leads to the uncontrolled immune response in the colon mucosa, and we examined the levels of TGF-β mRNA and of latent and active TGF-β1 protein. Although message was increased in colitis, very little difference was observed between control and diseased animals, using immunohistology or ELISA to assess levels of active or latent TGF-β1 protein. Latent protein was distributed throughout the mucosa, but the highest levels of active protein were associated with the basement membrane, with low levels observed in the gut LP; surprisingly, there were no obvious changes with disease. We also noted that the basement membrane matrix proteins, type IV collagen and laminin, were distributed throughout the LP in colitis. In this study we hypothesized that there would be changes in TGF-β receptor expression in the SCID mouse model that would account for the changes in mesenchymal cell ECM deposition associated with chronic inflammatory pathology and consequent epithelial barrier dysfunction.
Materials and Methods
Animals and Colitis Model
C.B-17+/+ mice (wild-type) and congenic C.B-17 scid/scid mice (control SCID) were bred and housed under specific pathogen-free conditions. Experiments conformed to local and national guidelines. Control and transplanted SCID (colitic SCID) mice were housed under identical conditions. Colitic SCID mice were monitored daily for signs of disease and weights were recorded every week. C.B-17 mice are congenic with Balb/c mice. The CD4+ T cell-transplanted C.B-17 SCID model of colitis was first described by Powrie et al. (1993). SCID mice were injected IP with 5 × 105 non-fractionated CD4+ splenic T-cells from wild-type congenic mice and monitored for signs of disease as previously described (Rudolphi et al. 1994). Samples were taken at autopsy 3–4 four months after transfer. Tissue was scored (1, mild to 3, severe) in a blinded fashion for each of the following six parameters of disease development: tissue hypertrophy; T-cell infiltration; crypt hyperplasia; crypt branching and distortion; crypt abscesses; and ulceration. Cumulative scores indicated level of pathology: 0–6, mild; 7–12, moderate; and 13–18, severe. Colon tissue samples were taken from five colitic SCID mice, five age- and sex-matched control SCID mice, and five wild-type mice. Colons were divided into three equal parts of approximately 3 cm and labeled proximal, mid-, and distal colon.
Human Samples
Samples from four control human colons taken at tumor resection and three Crohn's disease and two ulcerative colitis resection specimens were collected from Southmead Hospital, (Bristol UK) and Southampton General Hospital (Southhampton, UK). Tissues were collected with approval of local ethical committees.
Immunohistochemistry
Samples were placed on cork discs (RA Lamb; London, UK), covered with OCT (RA Lamb), snap-frozen in isopentane cooled over liquid nitrogen, and stored at −70C. Five-μm sections for immunohistochemistry (IHC) from all groups of mice were cut at −20C on to the same slide and air-dried. Sections were fixed in acetone at 4C for 10 min and then rehydrated in PBS for 10 min. Staining for RII, α-smooth muscle actin (SMA), plasminogen, or vimentin was enhanced by a 10-sec pretreatment in 50% (v/v) methanol/PBS. Nonspecific binding was blocked with 10% (v/v) normal goat serum (rat monoclonals) or 10% normal donkey serum (goat or rabbit polyclonal antibodies) in PBS for 1 hr at 20C, followed by an avidin/biotin block (Vector Laboratories; Peterborough, UK). To block endogenous mouse immunoglobulins when mouse primary antibodies were used, the MOM blocking kit (Vector) was used according to the manufacturer's instructions, with either the supplied biotinylated secondary or with isotype-specific fluorochrome-conjugated goat secondary antibodies (Southern Biotechnology; Birmingham, AL). Primary rat and mouse monoclonal or rabbit and goat polyclonal antibodies used are given in Table 1. Primary antibodies or isotype-matched control immunoglobulins were diluted in PBS and usually applied at 4C overnight. As a further negative control for RI and RII, primary antibody was incubated with a fivefold excess of immunizing peptide for 3 hr at 20C before application to the sections. Secondary antibodies were biotinylated goat anti-rat (Harlan–SeraLab; Crawley Down, UK) 1:200, donkey anti-rabbit (Stratech; West Grove, PA) 1:500, and donkey anti-goat (Stratech) 1:250. Where possible, multiple primary or secondary antibodies for dual or triple immunoflorescence were added together. However, when two biotinylated secondaries were used, one was applied first followed by streptavidin–FITC (Southern Biotechnology) (1:300), then a second avidin/biotin block, and then the second biotinylated secondary was added, followed by streptavidin–Texas red (TXRD) (Vector) (1:100), or avidin AMCA (Vector) (1:100). When two rabbit primary antibodies were used, the antigen giving the weakest signal under optimized conditions was incubated first with the relevant primary overnight and then developed with goat anti-rabbit (Fab fragment) conjugated with FITC (Stratech) (1:200). The second rabbit primary was then added to the sections and incubated at 20C for 1 hr and then incubated with biotinylated donkey anti-rabbit IgG (1:1000) for 1 hr followed by streptavidin–Texas red. Some slides were developed using ABComplexes (DAKO; Glostrup, Denmark) and peroxidase with DAB as substrate, as previously described (Whiting et al. 2001). Cells grown on chamber slides were washed three times in PBS, air-dried for 1 hr and then treated identically to tissue sections. All chemicals were from Sigma (St. Louis, MO) unless otherwise indicated.
Image Capture and Analysis
Images were viewed using a Leica DMRB microscope (Leica UK; Milton Keynes, UK) and grabbed using a Colour Coolview CCD camera (Photonic Sciences; Robertsbridge, E. Sussex, UK) and Image-Pro Plus software (Media Cybernetics; Baltimore, MD). Multiple fluorescent images were viewed using a triple-pass filter. Sensitivity within the red, green, and blue channels and other camera settings was set at the start of each of the six analyses of images and kept constant for all images captured. Six to eight stored images of non-ulcerated colon, from each colon region for each group, were then analyzed automatically using Image Pro-Plus software (Johnson et al. 2001) for μm2 area or short axis diameter for double-stained cells. Using two images from each group in each experiment, intensity ranges for double-stained cells were determined by defining the red, green, and blue (RGB) values for double-stained cells, which were at least twice background. RGB intensity ranged from 1 (no light) to 255 (maximal intensity), positive stain for both red and green, ranged from a minimum of 41–90 to a maximum of 255, and background was usually below 20. The RGB intensity range for double-stained cells was defined by the same investigator for each experiment and was therefore constant when applied to each image analyzed. The short axis diameter was defined as the length of the shortest line that can be drawn through the centroid joining two points on the object's perimeter, and was used as an estimate of cell size.
Primary antibodies
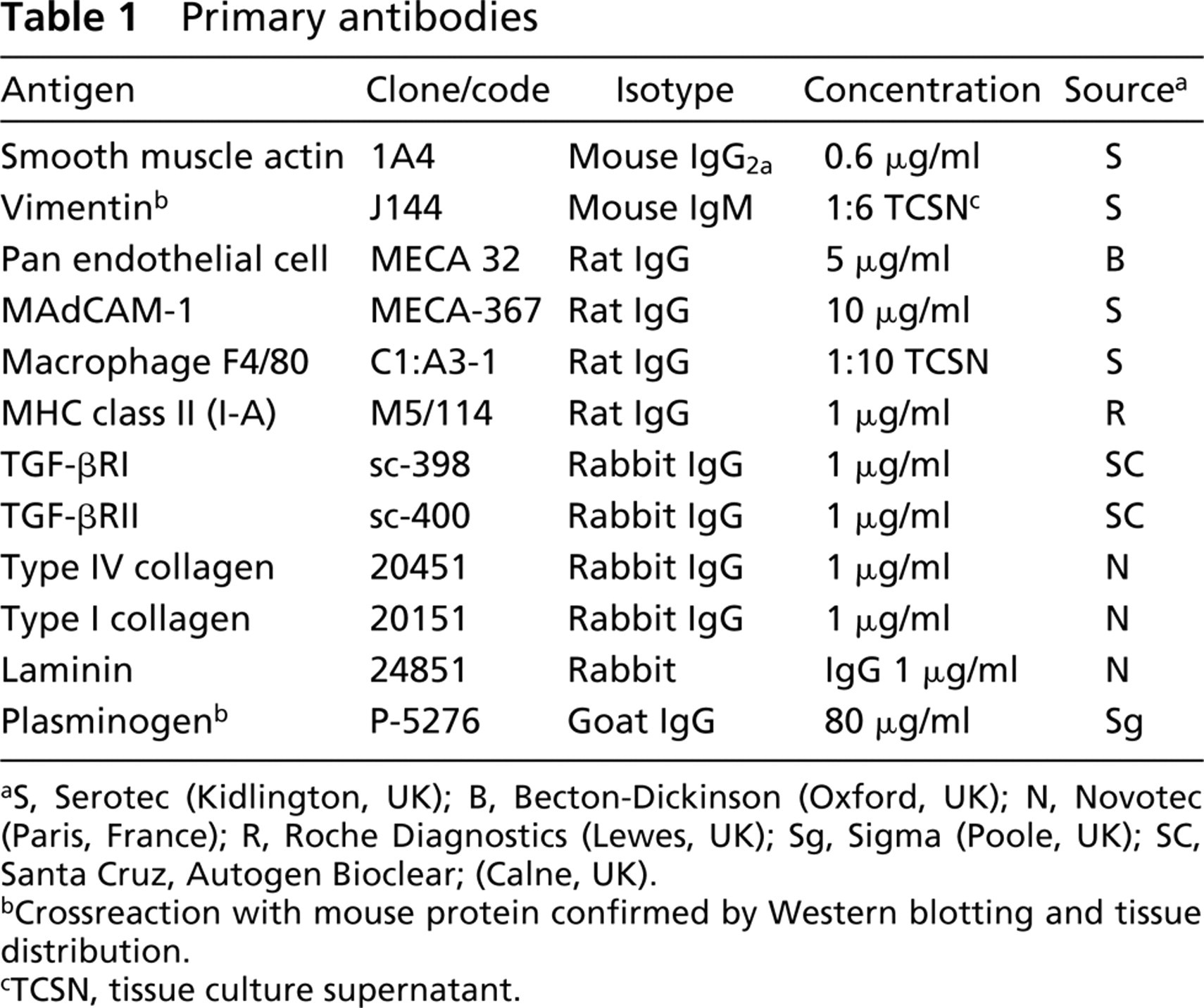
S, Serotec (Kidlington, UK); B, Becton-Dickinson (Oxford, UK); N, Novotec (Paris, France); R, Roche Diagnostics (Lewes, UK); Sg, Sigma (Poole, UK); SC, Santa Cruz, Autogen Bioclear; (Calne, UK).
Crossreaction with mouse protein confirmed by Western blotting and tissue distribution.
TCSN, tissue culture supernatant.
Measurements were made of the distance between SMA+ cells and the basement membrane in non-ulcerated colon. In six experiments, tissue sections from all groups of mice and colon regions were double stained for SMA and type IV collagen. Two mice from each group were randomly selected and then one image from each region was randomly selected for analysis, with a total of six images analyzed for each group. The image was magnified by two levels and a minimum of 10 cell: BM distances were determined by drawing lines along the proximal (to the nearest BM) cell surface and then along the epithelial surface of the BM opposite the same cell, for the length of that cell. Image analysis was then used to determine the mean distance between the lines and data for the images from each group were pooled.
Cell Culture
Colons were taken from three 6-week-old Balb/c mice, cut open, and vigorously washed in cold PBS. The epithelium and mucus were scraped off with a scalpel blade and discarded. The remaining tissue was minced with two scalpel blades in serum-free RPMI culture medium containing antibiotics (penicillin 200 U/ml, streptomycin 200 μg/ml, gentamycin 100 μg/ml, and amphotericin B 0.25 μg/ml) and HEPES (20 mM). The pieces were washed with two changes of medium. Six-well plates were seeded with one sixth of a colon per well in 1 ml α-MEM containing antibiotics, ribonucleosides, and 10% FCS and cultured in a humidified CO2 incubator. Half the medium was replaced after 4 days, and by day 6 outgrowing fibroblasts were observed. Cells were allowed to grow in six-well plates with weekly medium changes until about day 21, when cells were removed with trypsin–EDTA and placed in 25-cm2 flasks. Thereafter, cells were trypsinized when confluent and flasks reseeded at 100 cells/mm2 either in tissue culture flasks or chamber slides (Falcon). Each trypsinization was considered as an increase in passage number. All culture media and supplements were from Invitrogen (Glasgow, UK).
Statistics
For analysis of image data from tissue sections, differences in untransformed cell size or area measurements were determined by analysis of variance using mouse group, colon region, and mucosal site as factors. For the analysis of distance of cells from the basement membrane, analysis of variance was carried out on log-transformed measurement data followed by a post hoc analysis to determine the level of significance between groups. Results were considered significant at p≤0.05.
Results
Disease Scores
Tissues from all regions of inflamed SCID mouse colons were moderately diseased, with colon region scores ranging from 4 to 13 (average = 8.6/18 maximum), similar to those previously reported by our laboratory (Williams et al. 1999; Whiting et al. 2001).
TGF-β Receptor I and II Distribution in Wild-type, Control SCID, and Colitis SCID Mouse Colon
The distribution of RI was similar in wild-type (Figure 1A), control SCID (Figure 1B), and colitic SCID (Figure 1C) tissue. Subjectively, RI was expressed at high intensity by epithelial cells (Figures 1A–1C) and all muscle cells, including smooth muscle of the outer muscle layers (Figures 1A–1C), the muscularis mucosa (Figures 1B and 1C) and surrounding vessels (Figure 1B). Cells in the LP showed lower intensity expression. Epithelial RI was expressed to the same intensity along the length of the crypt (Figures 1A–1C). RII was also expressed at high intensity by all smooth muscle cells, including smooth muscle of the outer muscle layers and muscularis mucosa (Figures 1E and 1G) and surrounding vessels (Figure 1E), in all groups of mice. Epithelial cells along the length of the crypt expressed RII and, subjectively, the intensity of epithelial expression was lower than that observed on muscle cells. Many LP cells expressed low levels of RII, but scattered spindle-shaped cells expressed RII at a particularly high intensity in controls (Figures 1E and 1F) and colitic SCID (Figure 1G). The enlarged and elongated polymorphic cells were particularly prominent in colitis (Figures 1G and 1I). With high magnification, both RI and RII were observed throughout the cytoplasm of expressing cells but not in the nucleus (data not shown). The antibodies were raised against conserved intracellular epitopes, so the intracellular expression probably represents a combination of newly synthesized, endocytosed, and inner membrane proteins. The specificity of these antibodies, which has been reported previously (Jakowlew et al. 1998), was confirmed by Western blotting of tissue and fibroblast extracts where both anti-RI and anti-RII antibodies detected bands of the appropriate molecular weight (data not shown).
In summary, there was no change in RI expression with disease, but the large LP spindle-shaped cells seen in disease showed very high-intensity expression of RII. Our next series of experiments sought to phenotype this polymorphic TGF-β RII+ cell.
TGF-β RII Was Strongly Expressed Predominantly on Myofibroblasts and Fibroblasts
Double immunofluorescence was carried out to identify the spindle-shaped cells that expressed high levels of RII. Expression of RII on these cells was observed to mainly co-localize with SMA (Figure 2A) and plasminogen (Figure 2B). Lamina propria SMA also colocalized with vimentin (Figure 2D), an intermediate filament found in mesenchymal cells, indicating that most of these cells were myofibroblasts (MFB). As expected, vimentin was also expressed on all other LP cells, but epithelial and muscle cells were vimentin-negative. There were cells, or parts of cells, showing SMA expression alone, but because vimentin tended to be perinuclear both in vivo and in vitro (Figure 6F) this probably represented SMA staining of peripheral parts of MFB. However, the presence of SMCs in the mucosa in colitis cannot be discounted. A small population of RII+ cells not expressing SMA was observed in the submucosa and LP of all groups of mice, in colon mucosal lymphoid aggregates of wild-type mice, and in heavily infiltrated areas of diseased colon mucosa (Figure 2E). Using serial sections and double staining with combinations of antibodies against RII, SMA, type I collagen (CI) (Figures 2E–2G), and vimentin (data not shown), these cells were defined as RII+ CI+ vimentin+ SMA− fibroblasts expressing membrane RII and pericellular type I collagen. ECM proteins are rapidly secreted and deposited as insoluble fibers, so although cell-associated type I collagen was observed in tissue sections (Figure 5F) and in cultured cells (Figure 6D), some of the type I collagen staining represented matrix fibers. Because there is no marker for mouse fibroblasts, this population was further characterized to eliminate other cell types that may have co-localized with the spindle-shaped type I collagen deposition, but no co-localization was observed with immune cells, endothelium, or SMC (data not shown). To a lesser extent, RII also co-localized with endothelium (Figure 2H), MadCAM-1+ HEV (Figure 2I), F4/80+ macrophages (Figure 2K), and MHC class II+ (Figure 2L) cells. Although Figure 2I demonstrates quite high levels of RII expression by MadCAM-1+ HEV, such extensive co-localization was a relatively uncommon observation. The number of MadCAM-1+ vessels did increase in SCID mouse colitis (data not shown), but not to a level that could account for the number of RII+ spindle-shaped cells. The vessels were also morphologically quite distinct from MFB.
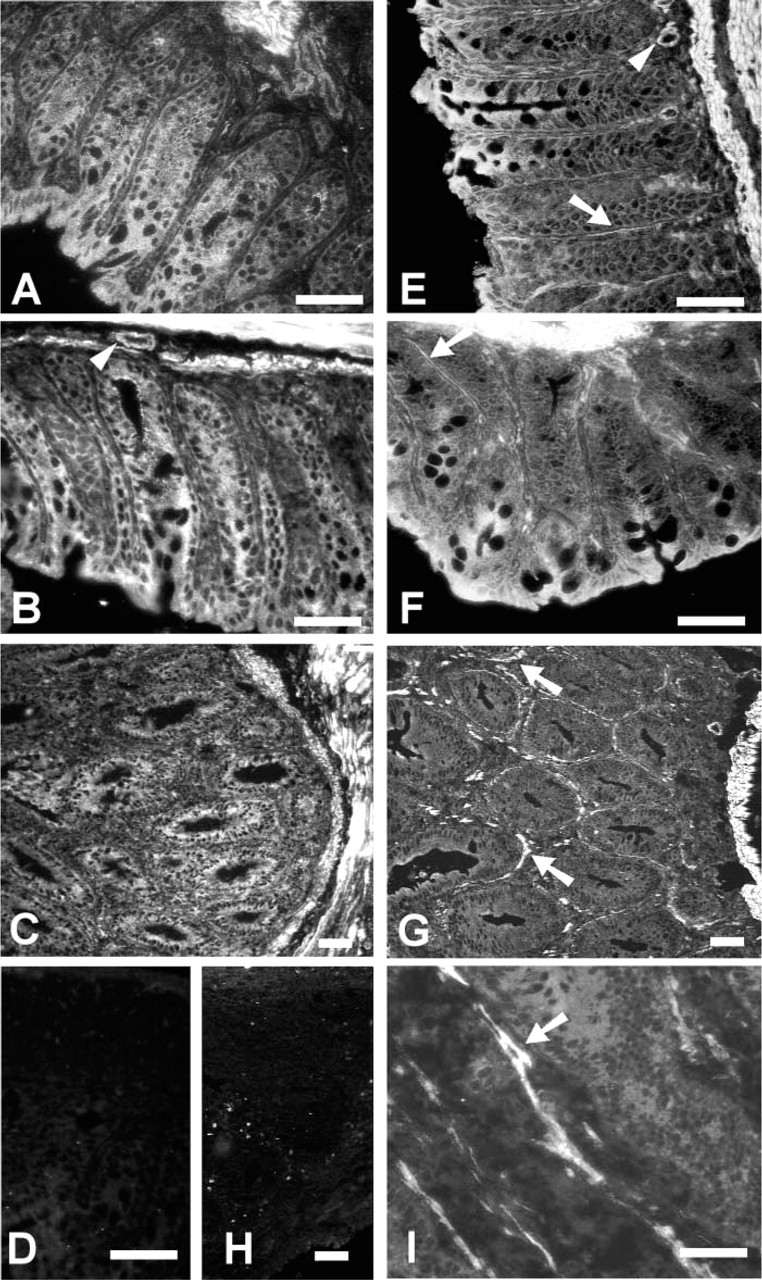
Distribution of TGFβRI (
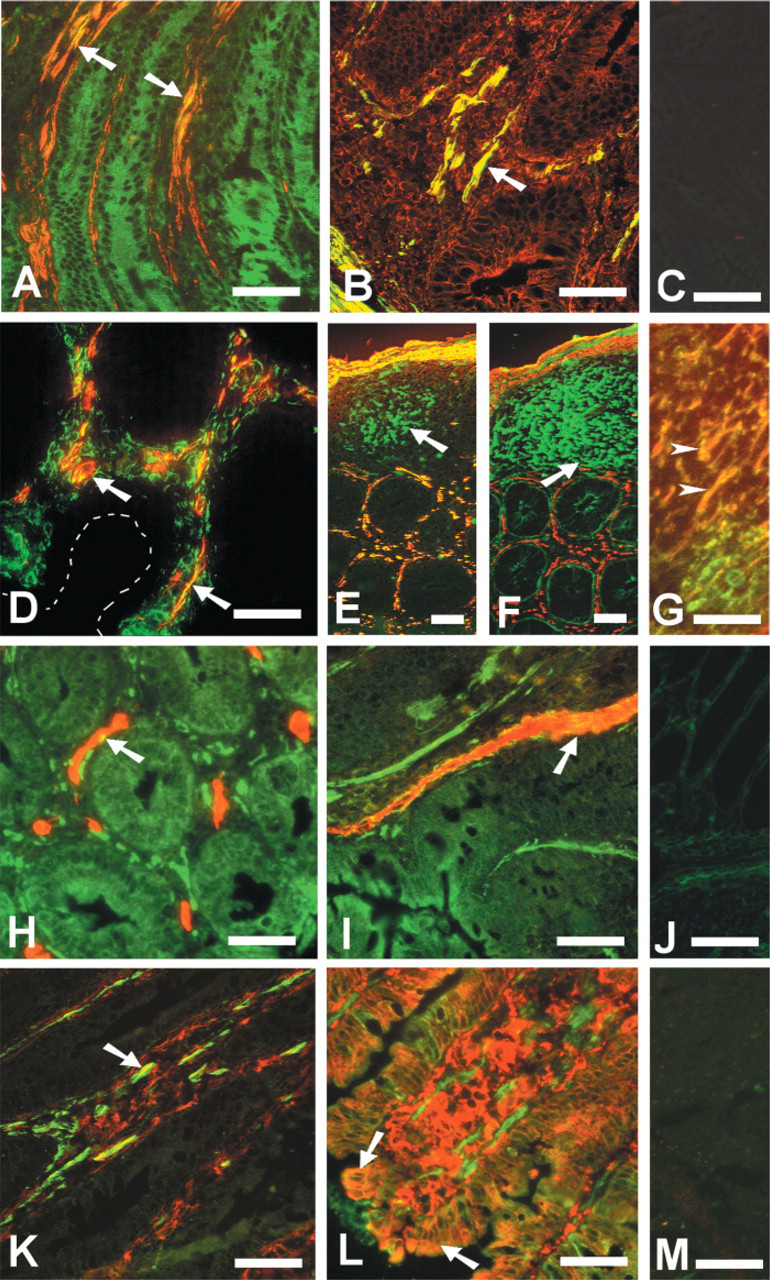
Lamina propria RII co-localizes mainly with myofibroblasts. Frozen sections of colitis tissue were stained by double immunofluorescence with Texas red- or FITC-conjugated antibodies. Co-localization is represented by yellow color. RII (FITC) co-localized with (
In summary, the major change in TGF-β receptor expression observed in colitis was the emergence of two populations of fibroblasts: (a) widespread, large LP myofibroblasts with an RII+ SMA+ vimentin+ plasminogen+ phenotype, and (b) fibroblasts with a RII+ SMA− vimentin+ type I collagen+ phenotype associated with mucosa, submucosa, and leukocyte aggregates in controls, and spreading to areas of intense inflammatory leukocyte infiltration in colitis.
RII+ SMA+ myofibroblasts increase in thickness and area in colitic SCID tissue. (
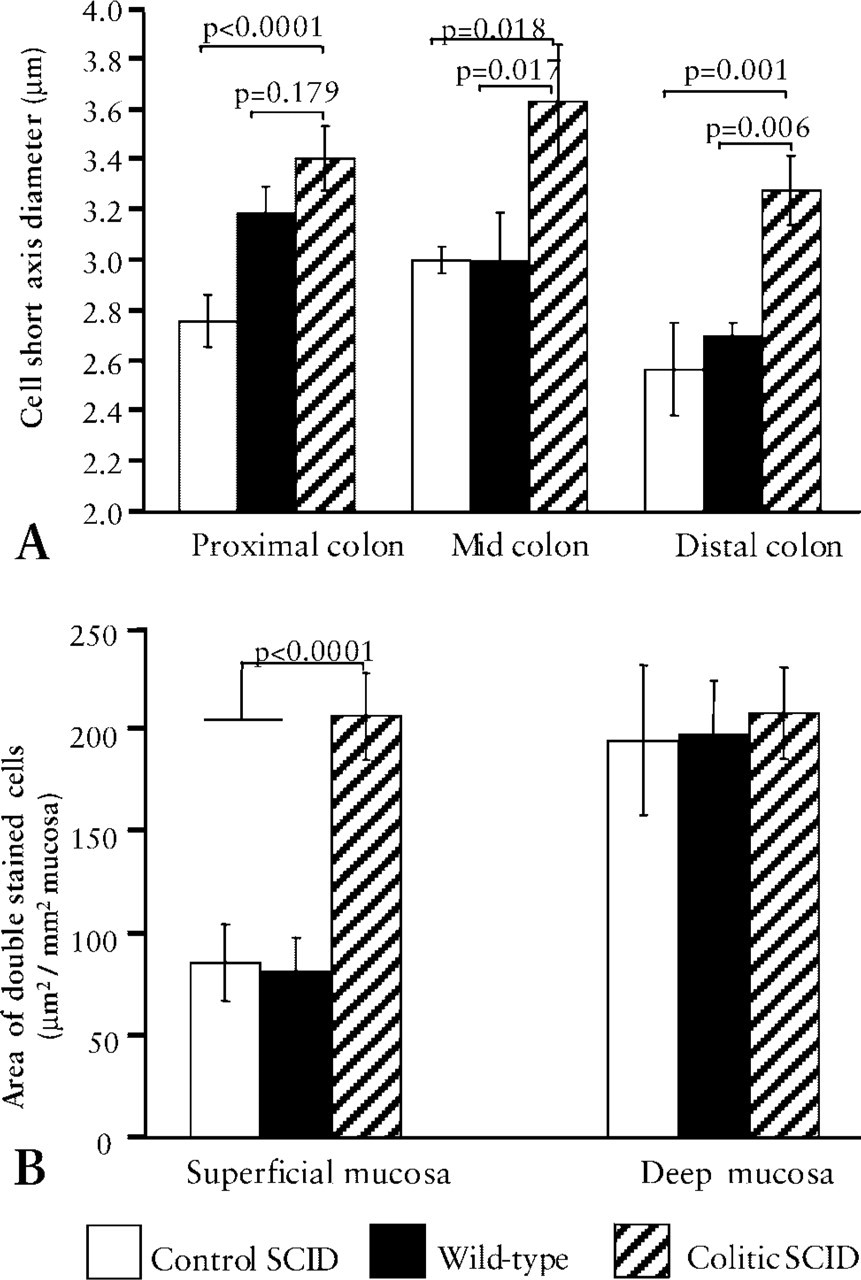
TGF-β RII+ Myofibroblasts Were Increased in Size and Occupied an Increased Mucosal LP in Colitis
It was not possible to count individual MFB, so short axis diameter of RII+ SMA+ cells was used as a measure of size, and area of mucosa occupied was used as a measure of the relative proportion of these cells in the tissue. Image analysis showed that, in inflamed tissue, RII+ SMA+ MFB were larger than in either of the control groups (Figure 3A), as determined by short axis maximal diameter. Mean cell size was greater in colitis in all regions of the colon, with 14/15 sections in the superficial mucosa and 10/15 sections in the deep mucosa having a mean short axis diameter larger than SCID control means (p<0.0001, p<0.02, and p = 0.001 for proximal, mid- and distal regions, respectively). Mean cell size was also greater in colitis than in wild-type controls in the mid- and distal regions (p<0.02 and p<0.01 respectively). Random orientation of these cells in the tissue precluded measurement of the long axis. In colitis, RII+ SMA+ cells were found throughout the mucosa in contrast to controls, in which they were mostly concentrated in the deep mucosa. There was a significantly greater area of superficial mucosa occupied by RII+ SMA+ cells in colitis compared to either of the control groups (p<0.0001 for both comparisons) (Figure 3B). In the superficial mucosa, an increased area of double-stained cells was observed in all regions of the colon, and 13/15 sections from colitic tissue had mean values greater than controls. The increased size and volume occupied by RII+ cells is particularly obvious in Figures 1G, and 1I vs 1E and 1F (note the scale difference).
In summary, the SMA+ RII+ LP MFB are enlarged and occupy an increased proportion of the mucosa in the inflamed colon.
SMA+ MFB Dissociate from the Basement Membrane in Colitis
Using image analysis on a random sample of colitis and control tissues, MFB were shown to spatially separate from the basement membrane (Figure 4). In control tissues, the median distance from the proximal edge of MFB, defined by SMA expression, to the epithelial surface of the BM was 1.85 μm (control SCID range 0.6–6.6 μm) and 2.3 μm (wild-type range 1.1–6.6 μm). The median distance observed in colitic SCID colon tissues (median 4.41) was significantly greater than both wild-type and SCID control tissue (p<0.0005 for both comparisons). There were many cells in colitic SCID colon that were close to the BM, but the range was greatly increased (1–17 μm). Wild-type control MFB were at a significantly greater distance from the BM than SCID control MFB (p = 0.033).
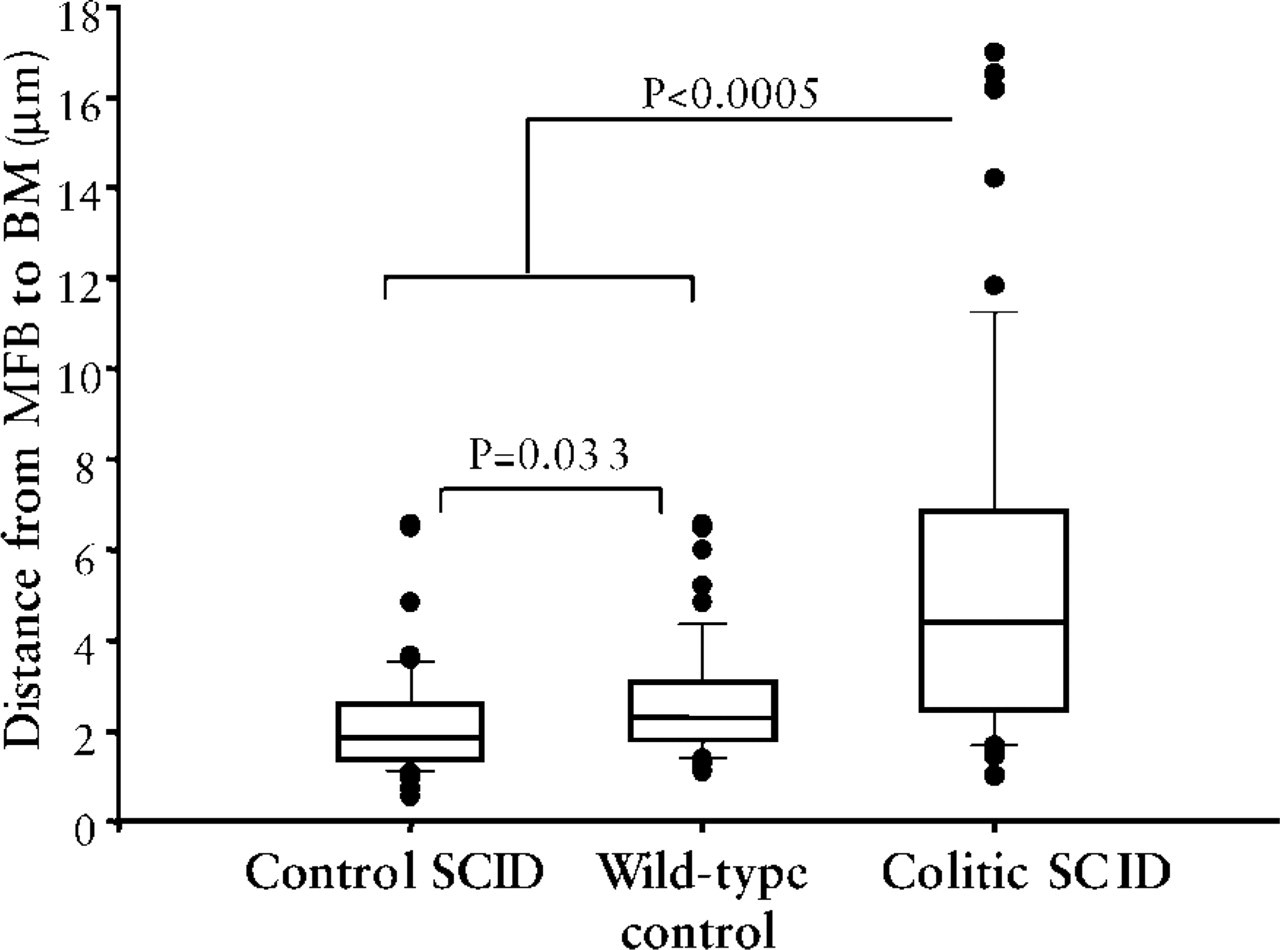
MFB spatially separate from the basement membrane in colitis. A random sample of control and colitis images covering all regions of the colon was analyzed. The distance from the surface of MFB nearest to the BM to the epithelial surface of the BM was measured as described in Materials and Methods on two mice per group. Data are plotted as a box and whisker plot, showing the median, 25th, and 75th percentile in the box. Whiskers are placed at the 90th and 10th percentile, and outliers are shown.
Changes in the ECM in Colitis
Myofibroblasts and fibroblasts contribute to ECM protein production and are responsible for all of the stromal type I collagen deposition in the mucosa. By double immunofluorescence, SMA+ MFB co-localized with both type IV collagen (Figures 5A–5C) and laminin (Figures 5D and 5E), two major BM proteins. Endothelium also co-localized with these BM proteins (Figures 5D and 5E), but there was no co-localization of laminin and type IV collagen with epithelial cytoplasm. Type I collagen also formed part of the BM, but SMA did not co-localize with either large spindle-shaped type I collagen or the dense bands of type I collagen observed in ulcers. This can be most clearly seen in Figure 2F, where type I collagen (labeled with FITC) is just visible surrounding each crypt but there was no co-localization of SMA (TXRD) with the type I collagen cluster. The anti-type I collagen antibody used did not crossreact with type IV collagen or laminin, as determined by ELISA (data not shown). Spindle-shaped cells expressing both type I and type IV collagens were observed in ulcer beds (Figure 5G, arrow). In wild-type and control SCID mouse tissue, the BM was thin and continuous, underlying the epithelium (Figures 5A, 5B, and 5D). However, in disease both type IV collagen (Figure 5C) and laminin (Figure 5E) were distributed throughout the LP and the BM was either more diffuse and occasionally thickened (Figures 5C and 5E) or was absent bordering crypt abscesses (Figure 5C). Occasionally, at the luminal surface (Figure 5F) and in ulcers (Figure 5G), normal BM, as defined by type IV collagen deposition, was absent or incomplete and was replaced by a dense band of type I collagen (insets in Figures 5F and 5G). In Figure 5F, cells expressing nascent, intracellular, or pericellular type I collagen (arrows, inset) can be seen just below the dense band of type I collagen that has been deposited in an area where the BM and epithelium appears to be dissociating from the underlying LP. The data suggested that there were at least two populations of mesenchymal cells synthesizing matrix within the mucosa. Although these results suggested that RII+ mucosal mesenchymal cells contribute to the de novo synthesis of laminin, type IV collagen, and type I collagen, it is also possible that co-localization simply indicates deposited ECM proteins surrounding or adhering to these cells. To investigate ECM synthesis by mucosal mesenchymal cells and to determine the relationship between SMA+ cells and ECM synthesis, we designed experiments using isolated mucosal cells.
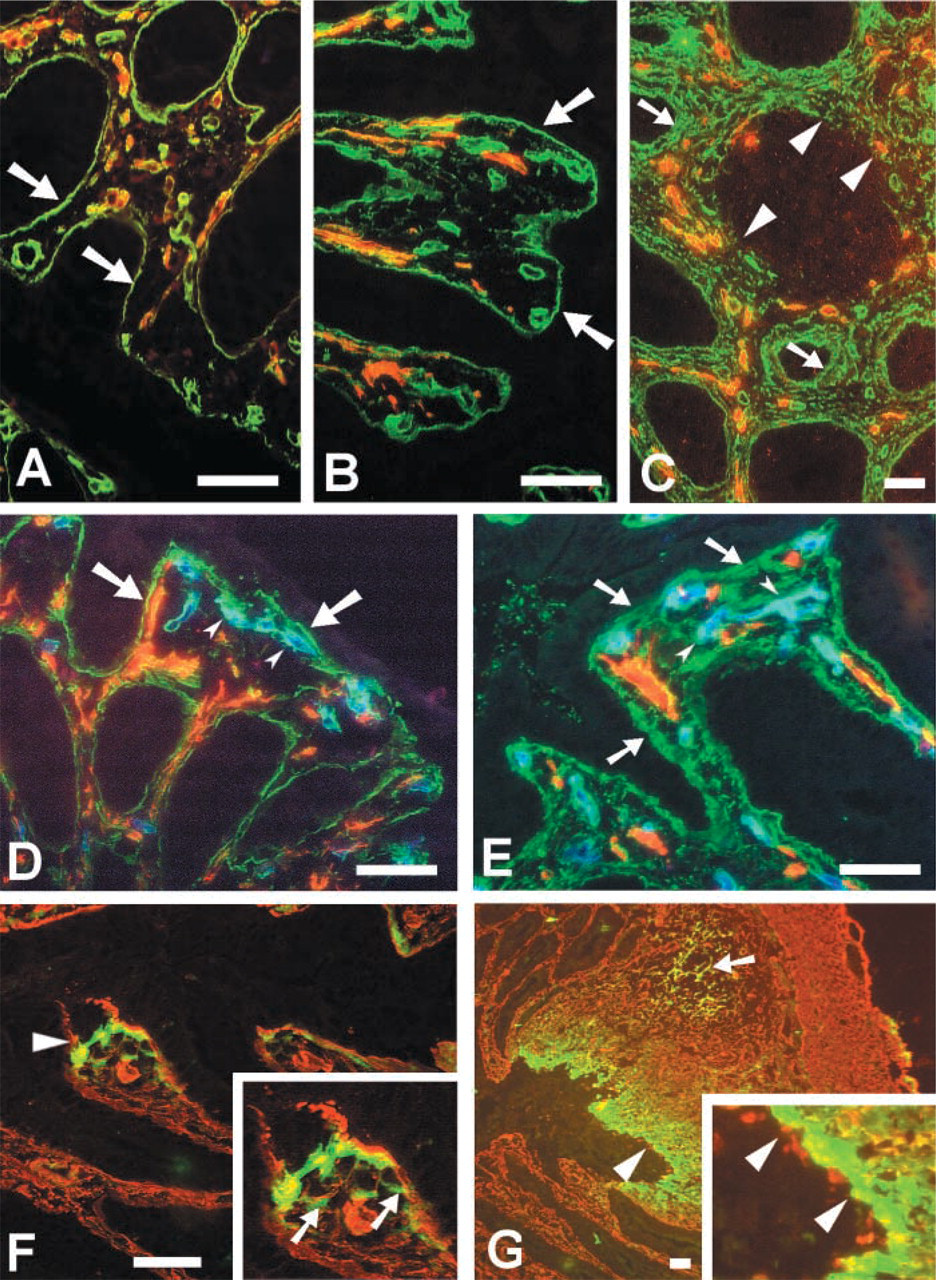
MFB co-localize with the basement membrane (BM) proteins, type IV collagen, and laminin, and, in colitis, BM protein distribution is disrupted. Matrix components were detected in frozen sections by double or triple indirect immunofluorescence using FITC-, TXRD-, or AMCA-conjugated antibodies. (
Ex Vivo Cultured MFB Have a Similar Phenotype to In Vivo Cells
Cultures of colon mesenchymal cells were established and analyzed for the expression of the same proteins as analyzed in frozen sections. Cells from passage 1 to 3 expressed vimentin, RI, RII, type IV collagen, and laminin (Figures 6B, 6C, and 6F–6H) but were a mixture of a-SMA positive and negative phenotype (Figure 6A) and exhibited variable type I collagen expression (Figure 6D). The majority of cells synthesized type I collagen, whereas only 30% of cells synthesized SMA at passage 1 (4 weeks of culture). Figures 6I and 6J show cells that expressed (a) both SMA stress fibers and type I collagen at passage 1 (arrows), (b) neither protein (open arrowheads), or (c) only type I collagen (closed arrowheads). Cells varied in their morphology, from fine spindles, which were usually SMA−, to large polymorphic cells that usually expressed SMA. Cultures did not contain endothelial cells or immune cells because there was no expression of either the panendothelial antigen or CD45. Further evidence that myofibroblasts and fibroblasts expressed functional TGF-β receptors was obtained from experiments in which the addition of TGF-β to cultured cells resulted in altered ECM synthesis (unpublished observations).
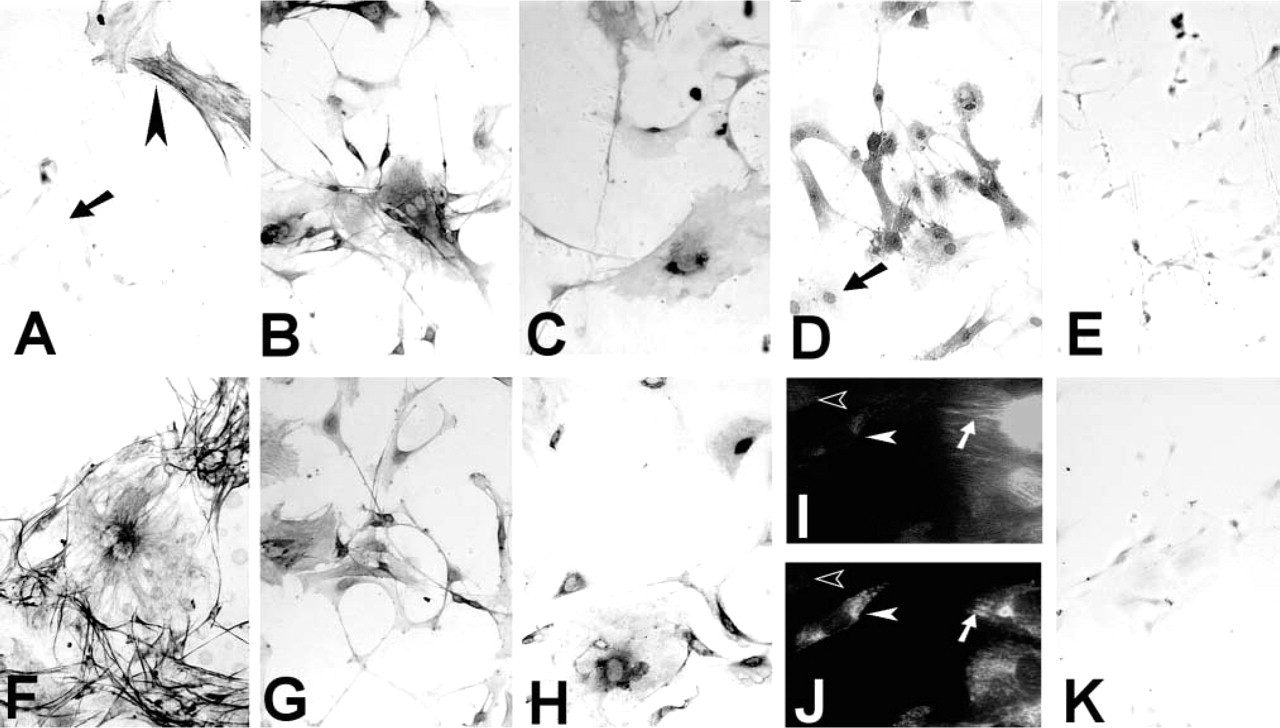
Characterization of isolated cultured myofibroblasts. (
RII-expressing MFB in Human Control and Colitis Tissue
In a limited study of five inflamed and four control human colons, SMA co-localized with RII expression on LP spindle-shaped cells in both controls (Figure 7A) and in colitis (Figure 7B). Figure 7 is representative of findings for all control and inflamed tissue studied. As observed in mouse colitis, RII+ SMA+ cells were greatly enlarged in colitis.
Discussion
Transforming growth factor-β has crucial roles in regulating both the immune response and ECM deposition in the intestine, and its activity is, therefore, tightly regulated. A major facet of this regulation is control of expression of the two main signaling receptors, RI and RII. This study shows that, in the colon, expression of these receptors in the normal state is concentrated on smooth muscle elements and the epithelium, where they presumably function in matrix homeostasis (Coutts et al. 2001) and differentiation (Halttunen et al. 1996), respectively. However, in colitis induced in SCID mice by the adoptive transfer of normal CD4+ T-cells, there is an expansion of activated LP mesenchymal cells expressing TGF-βRII. Our results for receptor distribution in normal colon broadly agree with previous studies in human and rat tissue (Winesett et al. 1996; Eskinazi et al. 1998; Planchon et al. 1999; Zhang et al. 1999). However, we detected no major changes in RI, whereas reports in fixed human Crohn's disease tissue (Di Mola et al. 1999) suggest that RI and RII, although not expressed in healthy mucosa, appear in disease. Although it has been inferred that mesenchymal cells contribute to matrix changes in gut inflammation (Mahida et al. 1997), their changed activity in mucosal inflammation has not previously been conclusively linked to regulation by TGF-β. Our studies identifying strong expression of TGF-βRII on mesenchymal cells, along with disordered ECM deposition, provide further evidence that TGF-β is involved in mediating connective tissue pathologies in colitis. Moreover, our preliminary results showing enlarged MFB expressing RII in human IBD are very similar to our observations in the mouse model.
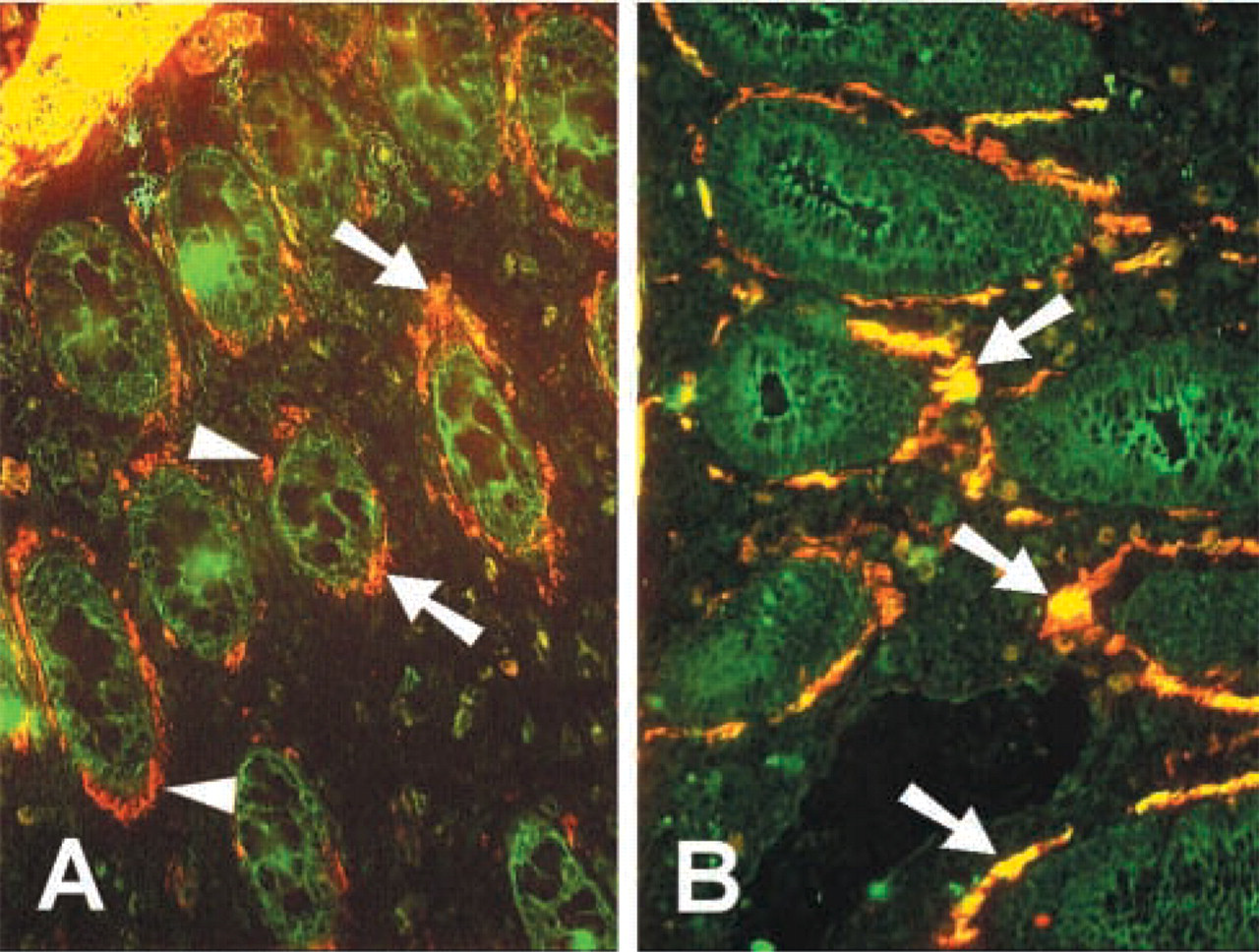
TGF-βRII expression co-localizes with SMA in human colon. Frozen tissue sections of control or colitis tissue were stained by double immunofluorescence for RII (FITC) and SMA (TXRD). (
It is perhaps surprising that, given the role of TGF-β in regulating colitis in this model (Powrie et al. 1996) and its proposed wider role in the homeostatic/inflammatory balance in the gut (Strober et al. 1997), we found only low RI and II expression by LP mononuclear cells, with significant expression only by MFB. Low-level signaling receptor expression may be a contributing factor to the maintenance of chronic inflammation. It is possible, however, that this low RI and RII expression is countered by co-expression of molecules that increase the affinity of RII for its ligand. Betaglycan (Zhang et al. 1999) and endoglin (St-Jacques et al. 1994) have been shown to be expressed in the gut LP, but preliminary studies in this laboratory have shown that betaglycan was expressed predominantly on endothelium.
Our observation that colon mucosal MFB also co-localized with plasminogen gives a clear indication that one of the most important mechanisms for the local activation of TGF-β is associated with these cells. Plasminogen, when cleaved by plasminogen activator to plasmin, is known to activate latent TGF-β, and its co-localization with RII makes it likely that these cells are able to generate and respond to active TGF-β. In further support of this, McKaig et al. (1999) showed that cultured human MFB produced bioactive TGF-β3, probably as a result of cell surface plasminogen activator receptor/plasminogen activator complex cleaving serum plasminogen to generate active plasmin.
Myofibroblasts were much enlarged in colitis, in all regions of the colon. It is likely that increased size of MFB equates with increased activation status. LPS-activated gut MFB exhibit increased proliferation and ECM synthesis and produce elevated levels of inflammatory mediators (Chakravortty and Kumar 1997,1999), while TNF-α-stimulated MFB produced increased levels of activated MMP (Pender et al. 1997). Previous work in our laboratory, using the SCID mouse model, demonstrated elevated TNF-α in areas of crypt destruction and tissue infiltration (Williams et al. 1999). It is likely, therefore, that MFB in colitis tissue are activated and contributing to the pro-inflammatory cycle. The observation that MFB become spatially separated from the BM may have consequences for cellular crosstalk and control between MFB and epithelial cells, which has been demonstrated in vitro (McKaig et al. 1999). The dissociation of MFB from the highest concentrations of TGF-β in the BM may result in reduced ECM synthesis.
Myofibroblasts are important in the intestine to help maintain barrier function and are also involved in the immune response. Our studies have shown that in vivo, and in isolated cells, MFB co-localize with the BM proteins type IV collagen and laminin. BM proteins have previously been detected in cultured human MFB (Mahida et al. 1997), and mesenchymal cells have been shown to be responsible for most of the BM protein synthesis in mouse/chicken chimeras (Kedinger et al. 1998). We have shown that BM proteins in colitis have a dysregulated distribution. Disrupted type IV collagen deposition and loss of laminin from the BM have been demonstrated in human UC (Schmehl et al. 2000), and Wheatcroft et al. (1999) showed evidence for denatured type IV collagen throughout the LP in late-stage disease. A consequence of this elevated and inappropriate deposition of BM proteins could be elevated retention, activation, protease secretion by, or survival of, immune cells (Bank et al. 1994; Khan and Falcone 1997) which, combined with loss of epithelial barrier integrity, maintains the chronic inflammatory cycle.
The deposition of type I collagen in areas in which laminin and type IV collagen have been lost probably represents attempted wound healing. In both healthy and inflamed intestine, type I collagen was deposited at a low level in the epithelial BM, similar to findings in the human colon (Aigner et al. 1997). Pericryptal type I collagen in the deep mucosa was probably produced by a mixed population of SMA+ and SMA− fibroblasts, but the subepithelial type I collagen in the surface mucosa and in ulcers was probably produced by SMA− fibroblasts. Others have noted SMA− fibroblast-like cells in ulcers (Pender et al. 2000), and only a proportion of cells expressing type I pro-collagen mRNA were SMA+ in collagenous colitis (Gumther et al. 1999) and experimental gastric ulcer (Shahin et al. 2001). It was also noted that SMA+ pericryptal fibroblasts occurred only in the lower two thirds of the normal human colon (Sappino et al. 1989). We observed cell bodies surrounded by type I collagen in subepithelial areas, and SMA+ cells were never observed in areas of dense deposition of type I collagen. Strong et al. (1998) isolated mesenchymal cells with variable SMA expression from human intestinal tissue and, at the first passage, type I collagen+ SMA− cells were observed in our experiments. Therefore, we believe that both fibroblasts and myofibroblasts are present in the colon. TGF-β promotes type I collagen transcription (van Tol et al. 1999) in mucosal fibroblasts. We propose that, should the epithelial barrier be breached, BM-associated active TGF-β (Whiting et al. 2001) would be liberated as a result of elevated local proteolytic activity (Tarlton et al. 2000) and would upregulate fibroblast type I collagen production to seal the wound, thus reducing further ingress of aggressive luminal contents. It is not known how replacement of BM proteins with interstitial type I collagen affects subsequent pathology or repair. It has been shown that adherence of epithelial cells is decreased, and migration increased, on type I collagen compared with basement membrane (Stenn and Depalma 1988) and that re-epithelialization occurs more rapidly if the BM is intact (Ashcroft et al. 1995).
To characterize the mesenchymal cell population for future analysis of the effects of TGF-β in vitro and to determine the relationship between SMA and type I collagen, we carried out an analysis of cell phenotype and matrix synthesis by ex vivo cultured mucosal MFB. Although the elevated deposition of BM proteins throughout the lamina propria could have been due to endothelium, only fibroblasts and myofibroblasts expressed high levels of TGF-βRII. The isolated cells represented a mixed population with a similar phenotype to the mesenchymal cells in the mucosa in tissue sections, i.e., all early-passage cells expressed vimentin, type IV collagen and laminin, in agreement with data for adult human MFB (Mahida et al. 1997), but expression of SMA was variable. TGF-βRII and RI were expressed by cultured cells, but only RII was clearly expressed by mucosal mesenchymal cells in tissue sections, indicating that RI expression is increased as a result of culture. The low level of RI expression on mucosal MFB compared with high expression on outer smooth muscle cells in vivo may indicate a variation in the signaling receptor ratio between the two cell types and may result in a different response to TGF-β (Geiser et al. 1992). TGF-β receptor expression by isolated gut MFB has not previously been reported, but these cells have been shown to respond to TGF-β in a rat model of gut fibrosis (van Tol et al. 1999). Expression of type I collagen by most cells at early passage, with some co-expression with SMA, is at odds with the in vivo data, in which SMA did not clearly co-localize with type I collagen. SMA+ cells could have deposited type I collagen while migrating through the tissue, and, at very high image magnification, a few pericryptal SMA+ MFB did express a very low level of type I collagen. It is well documented that culture conditions can induce changes in both SMA expression (Masur et al. 1996; Vaughan et al. 2000) and expression of surface proteins (Racine–Samson et al. 1997). Therefore, type I collagen synthesis may have been stimulated in these cells as a result of culture procedures.
TGF-β-mediated pathology in the colon will depend on the level of biologically active protein and the level of intracellular signaling after ligation of target cell receptors. MFB and fibroblasts certainly produce type I collagen and BM proteins, and their expression of high levels of TGF-βRII indicates that they are capable of responding to TGF-β in vivo, probably by upregulating matrix synthesis. It is probable that, in normal uninflamed intestine, MFB are in a quiescent state and BM protein synthesis is controlled by crosstalk with the epithelium, including control by TGF-β. MFB response to TGF-β could be altered on activation during inflammation, potentially losing the control of directed synthesis provided by the epithelium. Alternatively, due to tissue architecture disruption, MFB spatially separate from the high level of BM TGF-β and produce BM matrix in a non-directed fashion throughout the mucosa, resulting in decreased deposition at the BM. On the basis of the data, one of the consequences to disease pathogenesis of changed activation status and ECM production of the mesenchymal cell population is likely to be compromised epithelial barrier function.
Footnotes
Acknowledgements
Supported by European Union grant QLGI-CT-1999-00050.
We would like to thank Dr Jorg Reimann (Ulm University, Germany) for providing mouse tissue, and Prof Thomas T. MacDonald (Southampton University, UK) for human IBD tissue.