
Abstract
Enhanced GFP (EGFP) is a powerful tool for the visualization of tagged proteins and transfected cells and is easily detected by fluorescence microscopy or flow cytometry in living cells. However, soluble EGFP molecules can be lost if cell integrity is disrupted by freezing, sectioning, or permeablization. Furthermore, the fluorescence of EGFP is dependent on its conformation. Therefore, fixation protocols that immobilize EGFP may also destroy its usefulness as a fluorescent reporter. Here we determined which methods of preparing murine lymphoid tissues immobilized soluble EGFP protein and retained its fluorescence while simultaneously maintaining the antigenicity of various immunologically important molecules and best preserving the overall morphology of the tissues. We found that EGFP could not be visualized in frozen sections of spleen that had not been fixed before freezing. However, robust EGFP fluorescence could be observed in frozen sections of tissues fixed under various conditions. Fixation was important to immobilize EGFP rather than to maintain conformation, because only minimal EGFP could be detected by immunofluorescence in unfixed frozen sections. Although it had little effect on EGFP fluorescence, the inclusion of sucrose during fixation better preserved the morphology of fixed tissues. These methods also preserved the antigenicity of a wide variety of molecules used to identify cell types in lymphoid tissues.
G
Because the fluorescence of EGFP is independent of other proteins, co-factors, or substrates (Chalfie et al. 1994; Heim et al. 1994; Chalfie 1995) and can be used in viable cells, it is now the reporter molecule of choice in a variety of biological systems (Chiocchetti et al. 1997; Ikawa et al. 1998; De Giorgi et al. 1999; Cao et al. 2001). For example, EGFP is often recombinantly coupled to other proteins (Cao et al. 2001) and the location of the resulting fusion proteins is visualized in living cells using standard fluorescence microscopy or confocal fluorescence microscopy. EGFP is also commonly used in studies involving retroviral transfections (Persons et al. 1998; Barrette et al. 2000; Ramezani et al. 2000; Tahara-Hanaoka et al. 2002), in which the gene of interest and EGFP are simultaneously expressed as a bicistronic mRNA (Persons et al. 1998; Barrette et al. 2000; Ramezani et al. 2000; Tahara-Hanaoka et al. 2002). The inclusion of an internal ribosome entry site (IRES) element between the gene of interest and EGFP facilitates EGFP expression (Levenson et al. 1998; Metz et al. 1998; Jespersen et al. 1999; Chang et al. 2001; Abad et al. 2002). Transgenic mice that express EGFP under the control of ubiquitous promoters (Okabe et al. 1997) or cell-type specific promoters (Chiocchetti et al. 1997; Kawakami et al. 1999) are also popular, as are gene-targeted “knock-in” mice, which express EGFP under the control of an endogenous promoter (Mohrs et al. 2001).
The advantage of each of these systems is that the fluorescence of EGFP unambiguously tags recombinant proteins, marks transfected cells, or reports gene expression without steps that interfere with the integrity of the cells or that might introduce background or nonspecific fluorescence. Although the visualization of EGFP in living cells is sufficient for many procedures, other applications require the simultaneous detection of EGFP and other molecules. Unfortunately, soluble EGFP molecules are inefficiently retained in tissue sections or in cells that have lost membrane integrity (Chalfie 1998). Therefore, fixation is required before sectioning or permeabilization. However, because the fluorescence of EGFP is dependent on its conformation (Ormo et al. 1996; Brejc et al. 1997; Li et al. 1997), methods are required to maintain the conformation of EGFP in fixed tissues. Furthermore, these same fixation procedures must preserve the ability to detect other molecules by immunofluorescence and to maintain the architecture of the tissue. Here, we describe a method of processing murine secondary lymphoid tissues that allows us to track EGFP-expressing cells while simultaneously maintaining the antigenicity of many of the cell surface markers commonly used to define the architecture of secondary lymphoid tissues.
Materials and Methods
Mice
EGFP transgenic mice were generated as described (Wright et al. 2001). Ubiquitous EGFP expression in these mice is controlled by the chicken actin promoter and CMV enhancer using a modified version (Okabe et al. 1997) of the pCX vector (Niwa et al. 1991). The EGFP transgenic mice were back-crossed more than 10 times to the C57BL/6 genetic background. EGFP transgenic mice and C57BL/6 mice were maintained in the Experimental Animal Facility at Trudeau Institute. All procedures using animals were approved by the Trudeau Institute IACUC and were conducted according to principles outlined by the National Research Council.
Cell Transfers
Spleens were removed from EGFP transgenic mice and mechanically disrupted by passage through wire mesh. Single-cell suspensions were depleted of erythrocytes by incubation in 150 mM NH4Cl, 1 mM KHCO3, 0.1 mM EDTA, pH 7.3, for 5 min. The remaining leukocytes were washed and resuspended in 2% FCS in PBS. Recipient mice were injected IV with 1–5 × 107 splenocytes, depending on the experiment.
Fixation Procedures
For fixation by immersion, spleens were removed from recipient mice and cut in half. One half was saved for analysis by flow cytometry and the other half was fixed by immersion in either 4% paraformaldehyde or 4% formalin in PBS, with or without 7% picric acid and with or without 10% sucrose, as indicated in Table 1. The time of fixation varied from 30 min to 24 hr. For fixation by perfusion, mice were injected IP with 100 U heparin (Elkins-Sinn; Cherry Hill, NJ) and sacrificed by CO2 inhalation 20 min later. The thoracic cavity was opened and a 25-gauge needle was inserted into the right ventricle and connected directly to a 20-ml syringe for manual perfusion, or by 0.8-mm internal diameter Tygon tubing (BioRad; Hercules, CA) to a 20-ml syringe at a height of 45 cm for gravity perfusion. The inferior vena cava was cut to allow outflow of fixative. Mice were first perfused with 20 ml of 2 U/ml heparin in PBS. This was followed by a second perfusion with 20 ml of either 4% paraformaldehyde or 4% formalin in PBS, with or without 7% or 14% picric acid, as indicated in Table 2. Mice were perfused a final time with 20 ml of 10% sucrose in PBS.
Decalcification of NALT
Mice that had been fixed by perfusion were decapitated and the skin, lower jaw, and incisors of the upper jaw were removed. The remaining tissue was placed in a rotating 50 ml tube with 7% EDTA and 10% sucrose in PBS. Decalcification took place over several days and the EDTA solution was changed daily.
Embedding and Sectioning
Fresh or fixed spleens were frozen directly in Tissue-Tec OCT (Fisher; Pittsburgh, PA) above liquid nitrogen. Fixed and decalcified heads were equilibrated in OCT over time by placing the heads in a rotating 50 ml tube with graded concentrations of OCT in PBS (25%, 50%, 75%, and 100%). Heads were embedded in OCT and frozen over liquid nitrogen. Frozen tissue blocks were brought to −20C and 7-μm sections were cut and placed on poly-l-lysine-coated slides. Slides were dried at room temperature (RT) overnight, then either probed with antibodies or stored at −20C.
Effect of immersion method on EGFP fluorescence, antibody binding, and tissue morphology
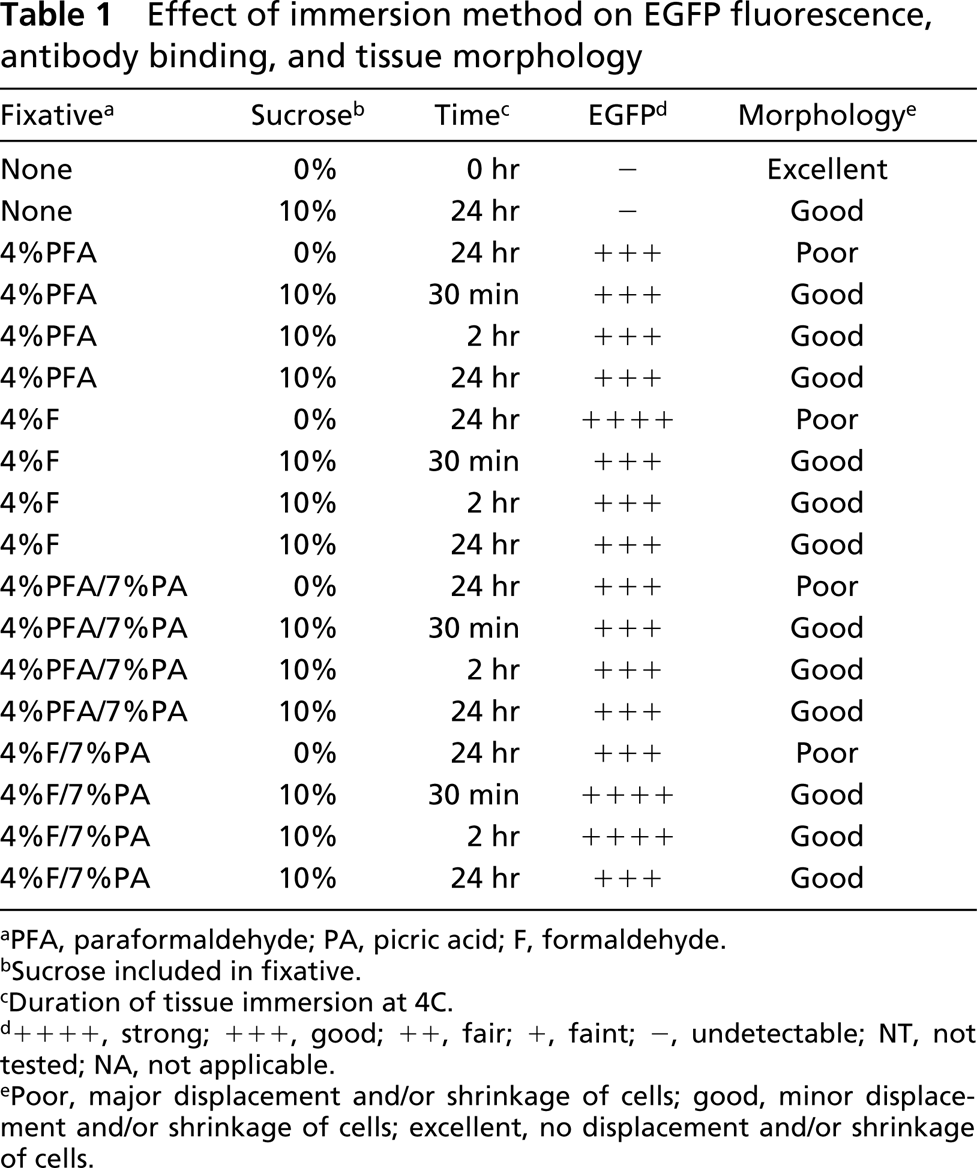
aPFA, paraformaldehyde; PA, picric acid; F, formaldehyde.
bSucrose included in fixative.
cDuration of tissue immersion at 4C.
d++++, strong; +++, good; ++, fair; +, faint; -, undetectable; NT, not tested; NA, not applicable.
ePoor, major displacement and/or shrinkage of cells; good, minor displacement and/or shrinkage of cells; excellent, no displacement and/or shrinkage of cells.
Effect of perfusion method on EGFP fluorescence, antibody binding, and tissue morphology
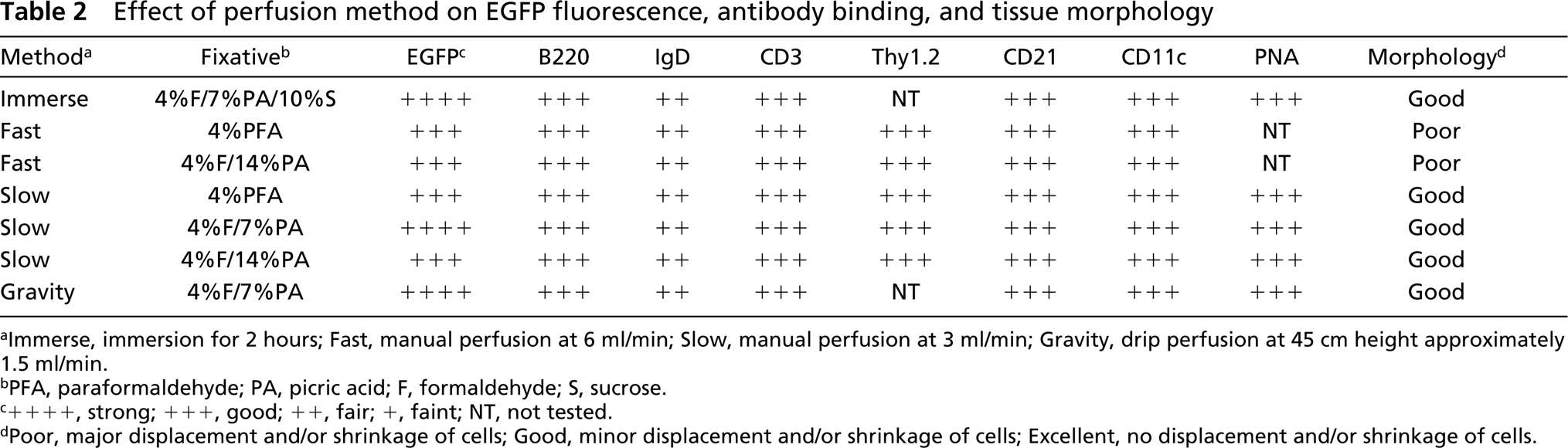
aImmerse, immersion for 2 hours; Fast, manual perfusion at 6 ml/min; Slow, manual perfusion at 3 ml/min; Gravity, drip perfusion at 45 cm height approximately 1.5 ml/min.
bPFA, paraformaldehyde; PA, picric acid; F, form aldehyde; S, sucrose.
c++++, strong; +++, good; ++, fair; +, faint; NT, not tested.
dPoor, major displacement and/or shrinkage of cells; Good, minor displacement and/or shrinkage of cells; Excellent, no displacement and/or shrinkage of cells.
Immunofluorescence
Slides were brought to RT and fixed in acetone at 4C for 10 min. Slides were then placed in PBS for 5 min to remove the OCT. All slides were blocked with 5% BSA in PBS for 30 min. Endogenous avidin and biotin were blocked with the Avidin-Biotin Blocking kit from Vector (Burlingame, CA). Slides were washed three times for 5 min each in PBS before being probed with biotinylated antibodies to various antigens for 30 min in a humidified chamber. Biotinylated anti-B220 (RA3–6B2), anti-CD3 (145-2c11), anti-Thy-1.2 (53-2-1), anti-CD11c (HL3), anti-CD21/CD35 (8C12), and anti-IgD (217-170) were purchased from PharMingen (San Diego, CA). Biotinylated peanut agglutinin (PNA) was obtained from Vector. Alexa 594-conjugated rabbit anti-GFP was purchased from Molecular Probes (Eugene, OR). After incubation with biotinylated antibodies, slides were washed three times for 5 min in PBS. They were then incubated with streptavidin-Alexa 594 (Molecular Probes) for 30 min, washed three times for 5 min each in PBS, and mounted with Polymount (Polysciences; Warrington, PA). In some experiments, sections were mounted with SlowFade Light antifade kit containing DAPI (Molecular Probes).
Microscopy and Digital Imaging
Slides were viewed with a Zeiss Axioplan 2 microscope using a 480/30 bandpass filterset to view EGFP, a 560/40 bandpass filter set to view Alexa 594, and a 330/80 filter set to view DAPI. Images were recorded with a Zeiss AxioCam digital camera (Zeiss; Thornwood, NY) using the Zeiss proprietary software, Axiovision 3.0.6.0. Images were manipulated in Adobe Photoshop 5.5 and saved as final figures in TIFF format at 600 DPI.
Flow Cytometry
In experiments in which spleens were not fixed by perfusion, the engraftment of EGFP transgenic cells into recipient spleens was monitored by flow cytometry. Half spleens were disrupted by passage through wire mesh and single-cell suspensions were obtained. Erythrocytes were lysed by 5-min incubation in 150 mM NH4Cl, 1 mM KHCO3, 0.1 mM EDTA, pH 7.3, and the remaining leukocytes were washed and resuspended in 2% FCS in PBS with 1 μg/ml propidium iodide. Cells were analyzed using a FACSCalibur (Becton-Dickinson; Mountain View, CA) flow cytometer. Dead cells were excluded from analysis by the fluorescence of propidium iodide in the FL3 channel and EGFP-expressing cells were identified in the FL1 channel.
Results
To determine if the fluorescence of EGFP-expressing cells was retained in frozen sections, we transferred splenocytes from EGFP transgenic mice to nontransgenic recipients. EGFP transgenic mice express EGFP constitutively in all tissues under the control of the chicken actin promoter and CMV enhancer. All leukocytes in these mice express uniformly high levels of EGFP and are easily distinguished from nontransgenic cells by FACS (not shown). Recipients were sacrificed 2 days after transfer and spleens removed for analysis. Half of each spleen was snap-frozen in OCT medium over liquid nitrogen, while the remaining tissue was mechanically disrupted and made into a single cell suspension for analysis by flow cytometry. Frozen tissues were sectioned (7 μm), air-dried, and fixed in acetone. Sections were then probed with biotinylated anti-B220 antibodies followed by streptavidin-Alexa 594 and analyzed by fluorescence microscopy. As shown in Figure 1A, whereas B220-expressing cells could be easily detected (red), no EGFP-expressing cells could be observed in snap-frozen sections, even though a substantial population of EGFP-expressing cells could be observed by flow cytometry (Figure 1A, inset). Because it was previously suggested that slow freezing of tissues better preserved EGFP fluorescence in frozen sections (Shariatmadari et al. 2001), we repeated the previous experiment but insulated the OCT-embedded tissue with cotton balls before freezing at −70C. Again, however, no EGFP expression was detectable (Figure 1B), even though EGFP-expressing cells were clearly present in the tissue as visualized by flow cytometry (Figure 1B, inset).
We next tested whether a cryopreservative, such as sucrose, maintained EGFP fluorescence in frozen sections. Therefore, we adoptively transferred EGFP-expressing cells into nontransgenic recipients for 2 days, immersed recipient spleens in 10% sucrose at 4C overnight, and embedded tissues in OCT by snap-freezing above liquid nitrogen. Sections from frozen tissues were probed with anti-B220 and analyzed by fluorescence microscopy. Again, no EGFP expression was detectable (Figure 1C), even though EGFP-expressing cells were clearly present in the tissue as visualized by flow cytometry (Figure 1C, inset).
We reasoned that the failure to detect EGFP expression was due either to the instability of the EGFP fluorophore in sections of frozen tissues or to the loss of the soluble EGFP from the sectioned cells. Therefore, to stabilize the fluorophore and to immobilize the soluble EGFP within the cells, we tried various methods of fixation before sectioning and analysis. As before, we transferred splenocytes from EGFP transgenic mice into nontransgenic recipients. Mice were sacrificed 2 days after transfer and spleens were removed and cut in half. Half of each spleen was mechanically disrupted and analyzed by flow cytometry to demonstrate the presence of EGFP-expressing cells, and the remainder of the tissues was fixed by immersion for 30 min, 2 hr or 24 hr at 4C in either 4% paraformaldehyde or 4% formalin in PBS, with or without 10% sucrose as a cryopreservative and with or without 7% picric acid as an additional fixative. Fixed tissues were then embedded in OCT and snap-frozen over liquid nitrogen. Frozen tissues were sectioned (7 μm), fixed in acetone, and probed with anti-B220 before analysis by fluorescence microscopy. As shown in Figure 1D, EGFP-expressing cells could be clearly observed in sections of tissues that had been fixed for 24 hr in 4% paraformaldehyde in PBS. The addition of 10% sucrose to this fixation protocol had little effect on EGFP fluorescence (Figures 1E and 1F). Similarly, EGFP-expressing cells could be observed in sections of tissues fixed for 24 hr in 4% formalin in PBS (Figure 1G). Again, the addition of 10% sucrose did not dramatically affect EGFP fluorescence (Figures 1H and 1I). Finally, EGFP-expressing cells could be observed in sections of tissues fixed for 24 hr with the combination of 4% formalin and 7% picric acid (Figure 1J), regardless of whether or not sucrose was added (Figures 1K and 1L). Therefore, fixation by at least three different methods preserved the fluorescence of EGFP in frozen sections.
Although fixation preserved the fluorescence of EGFP, the morphology of the fixed tissues (Figures 1D and 1L) was different from that of tissues frozen directly (Figures 1A-1C). Whereas cells in sections of unfixed spleens were plump and tightly packed (Figures 1A-1C), cells in sections of fixed spleens were somewhat desiccated in appearance and there were gaps between the cells (Figures 1D, 1G, and 1J). The addition of sucrose during the fixation process partially alleviated this problem (Figures 1E, 1H, and 1K). In particular, the addition of sucrose helped maintain cell shape (shown at higher magnification in Figures 1F, 1I, and 1L), even though there were still gaps between the cells.
To determine whether soluble EGFP was simply lost from sections of unfixed tissues or whether the protein was retained in a conformation that precluded fluorescence, we probed frozen sections of unfixed tissues with antibodies to GFP. As shown in Figures 1M and 1N, EGFP was barely detectable with anti-GFP antibodies in sections of tissues that had been snap-frozen (Figure 1M) or slow-frozen (Figure 1N), even though EGFP-expressing cells were clearly present by FACS analysis (insets). However, anti-GFP antibodies clearly detected EGFP in frozen sections of tissues previously fixed in 4% formalin/7% picric acid/10% sucrose (Figure 1O). In fact, all cells expressing EGFP were also detected by the anti-GFP antibody (Figure 1O, inset). Therefore, we conclude that the primary reason for loss of EGFP fluorescence in unfixed frozen sections is that soluble EGFP molecules are inefficiently retained in sections of tissues that are not fixed before freezing.
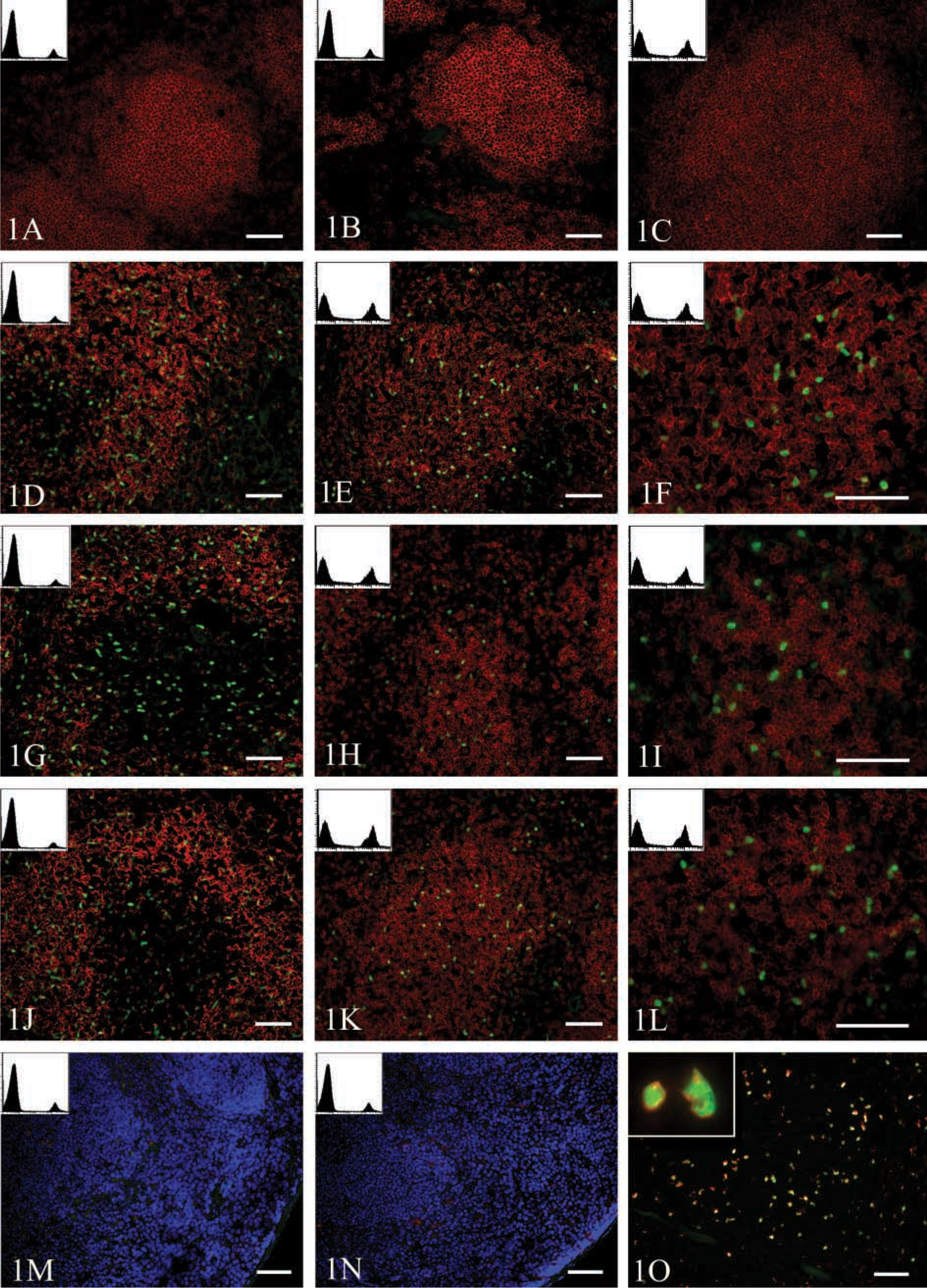
Fixation is required for the visualization of soluble EGFP in tissue sections. EGFP-transgenic splenocytes were adoptively transferred to nontransgenic recipients. After 2 days, recipient mice were sacrificed and spleens removed for analysis by flow cytometry and fluorescence microscopy. Half of each spleen was prepared by (
The results from the above experiments are summarized in more detail in Table 1. As discussed above, the presence and fluorescence of EGFP were maintained by all fixatives (Table 1), although we felt that the combination of 4% formalin and 7% picric acid in PBS consistently provided the brightest EGFP signal. Furthermore, the inclusion of sucrose consistently improved the morphology of the tissue, even though it had little effect on EGFP fluorescence (Table 1). Finally, the timing of fixation had little effect on morphology or EGFP fluorescence, although extended periods of fixation tended to compromise the binding of some antibodies, particularly anti-IgD (not shown).
To clarify whether the fixation conditions that best preserved both EGFP fluorescence and cell morphology also maintained the ability of various monoclonal antibodies to recognize their respective antigens on frozen sections, we transferred splenocytes from EGFP transgenic mice to a nontransgenic recipient and after 2 days we prepared half of the spleen for analysis by flow cytometry and fixed the remainder in 4% formalin, 7% picric acid, 10% sucrose in PBS for 2 hr before freezing, sectioning, and analysis by immunofluorescence. As shown in Figure 2, this fixation procedure maintained robust EGFP fluorescence and preserved the ability of multiple antibodies to bind to fixed sections. For example, anti-CD3 could be used to detect discrete T-cell areas in the spleen (Figure 2A). As shown at higher magnification in Figure 2B, many of the transferred EGFP-expressing splenocytes also expressed CD3 and were found in the T-cell area. Similarly, rounded B-cell follicles were observed with anti-B220 (Figures 2C and 2D) and with anti-IgD (Figures 2E and 2F). Furthermore, many of the transferred EGFP-expressing splenocytes also expressed B220 or IgD and migrated to the B-cell follicles (Figures 2D and 2F). Reticular networks of follicular dendritic cells (FDCs) could also be found within the B-cell follicles as visualized with anti-CD21 (Figures 2G and 2H). Although transferred EGFP-expressing cells were found adjacent to FDCs, none of the FDCs themselves expressed EGFP (Figure 2H), consistent with their stromal rather than hematopoietic origin (Matsumoto et al. 1997). CD11c-expressing interdigitating dendritic cells are normally found in the T-cell areas and in the bridging channels between the B-cell follicles (Steinman et al. 1997). We observed CD11c-expressing cells as reticular cells in the T-cell area (Figures 2I and 2J), although relatively few CD11c-expressing cells also expressed EGFP. Finally, we observed some clusters of PNA-binding germinal center B-cells (Figures 2K and 2L), none of which were of donor origin (Figure 2L). Therefore, this method preserved the fluorescence of EGFP and simultaneously maintained the antigenicity of a number of immunologically important molecules.
We next tested whether fixation by perfusion, rather than immersion, resulted in better EGFP fluorescence, antibody binding, and morphology. Therefore, we transferred EGFP-transgenic cells to non-transgenic recipients and sacrificed the recipients 2 days later. After sacrifice, mice were perfused through the right ventricle with 20 ml of heparin in PBS, followed by 20 ml of either 4% paraformaldehyde or formalin in PBS, with or without picric acid at various concentrations, as indicated in Table 2. Mice were finally perfused with 20 ml of 10% sucrose in PBS. The spleens were then excised, frozen in OCT over liquid nitrogen, and sectioned. As shown in Table 2, the fluorescence of EGFP was maintained in all of the perfusion conditions tested, although it was somewhat brighter in mice perfused with 4% formalin and 7% picric acid, similar to what we observed with the immersion fixation method (Table 1). Although the rate of perfusion did not affect the fluorescence of EGFP (Figure 3, insets), it markedly affected the morphology of the tissue (Figure 3). Rapid manual perfusion tended to displace B-lymphocytes from the follicles and to scatter them throughout the splenic matrix (Figure 3A). This was observed to a lesser extent with slow manual perfusion (Figure 3B) and was minimized by gravity perfusion (Figure 3C). However, even the gentle perfusion pressure induced by gravity resulted in cell displacement compared to immersion-fixation (Figure 2C) or snap-freezing (Figures 1A-1C). Again, the inclusion of sucrose maintained cell shape (Figure 3).
We also tested the ability of various antibodies to bind frozen sections of perfusion-fixed spleens. As shown in Table 2, all of the tested antibodies were able to detect their respective antigens on sections from spleens fixed under each of the tested conditions. This was somewhat surprising, considering that we had difficulty detecting IgD in sections of spleens fixed by immersion with similar reagents (not shown). However, other experiments suggested that the epitope recognized by the anti-IgD antibody is destroyed by longer fixation times. Therefore, the difference between the immersion-fixation and perfusion-fixation experiments is likely due to the length of time that the tissue is fixed rather than to the reagents used. Because tissues are in contact with fixative for less than an hour during perfusion, the epitope recognized by the anti-IgD antibody is maintained.
We next determined whether EGFP fluorescence was maintained through the prolonged process of fixation and decalcification required to section nasal associated lymphoid tissue (NALT) (Harmsen et al. 2002). Because NALT is directly adjacent to portions of the skull, decalcification of the bone is necessary to section this tissue. Therefore, we transferred cells from EGFP-transgenic donors to nontransgenic recipients and sacrificed the recipients 2 days later. Tissues were fixed by whole body gravity perfusion with 4% formalin and 7% picric acid in PBS, followed by 10% sucrose in PBS. Heads were removed and decalcified in 7% EDTA, 10% sucrose in PBS for several days before embedding in OCT and freezing. Sections were probed with anti-B220 and examined by fluorescence microscopy, as shown in Figures 4A and 4B. Transferred EGFP-expressing cells and B220-expressing B-cells could be easily observed in the mucosal lymphoid follicle just under the epithelial layer (Figure 4B and inset). Therefore, EGFP is retained in fixed tissues even through the prolonged process of decalcification necessary to section NALT.
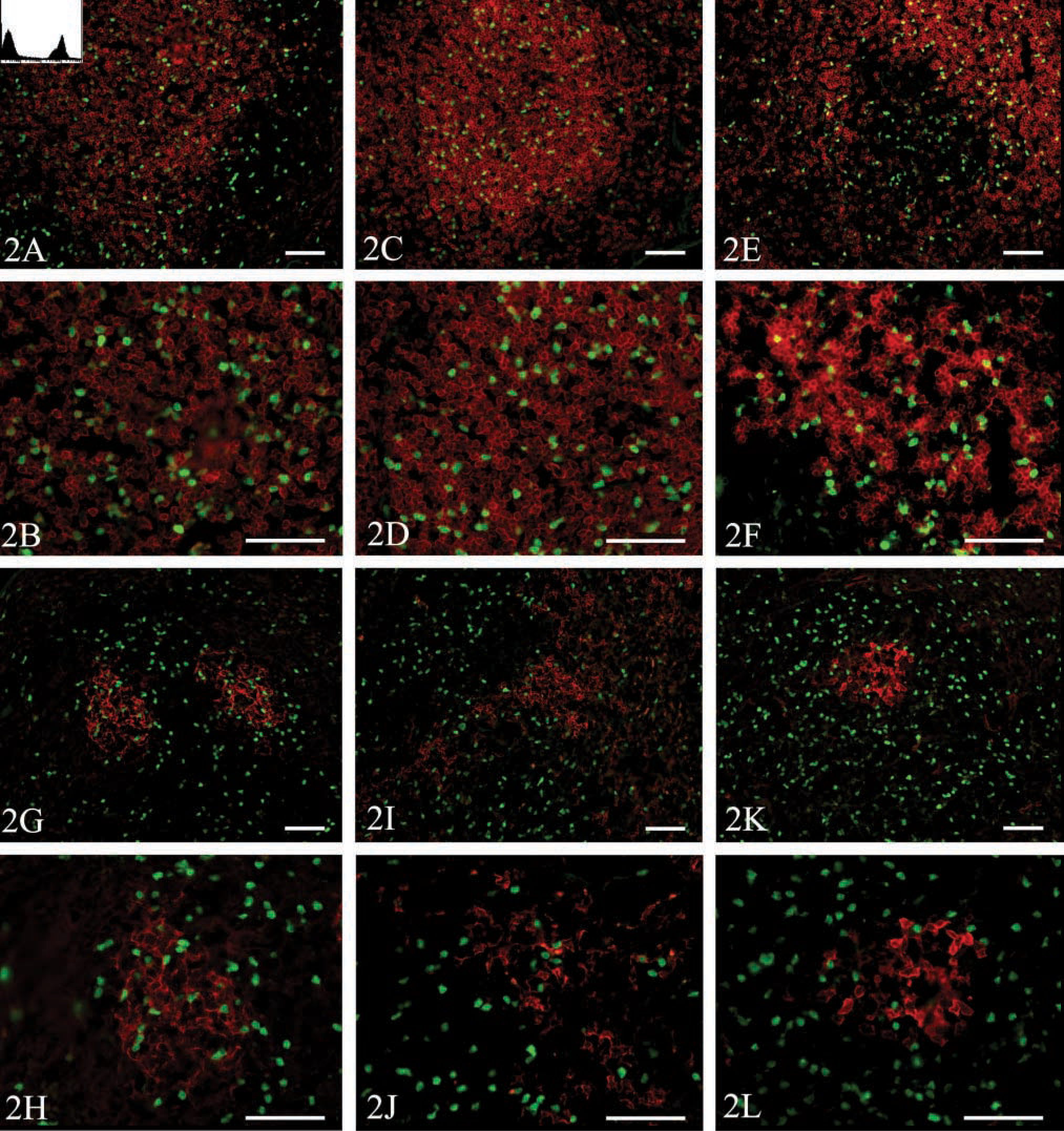
Analysis of antibody binding to fixed sections. EGFP-transgenic splenocytes were adoptively transferred into a nontransgenic recipient. After 2 days, the recipient mouse was sacrificed and the spleen was removed for analysis by flow cytometry and fluorescence microscopy. Half of the spleen was fixed in 4% formalin, 7% picric acid, and 10% sucrose in PBS for 2 hr and then embedded in OCT and snap-frozen over liquid nitrogen. Tissues were sectioned (7 μm) and were probed with biotinylated antibodies against (
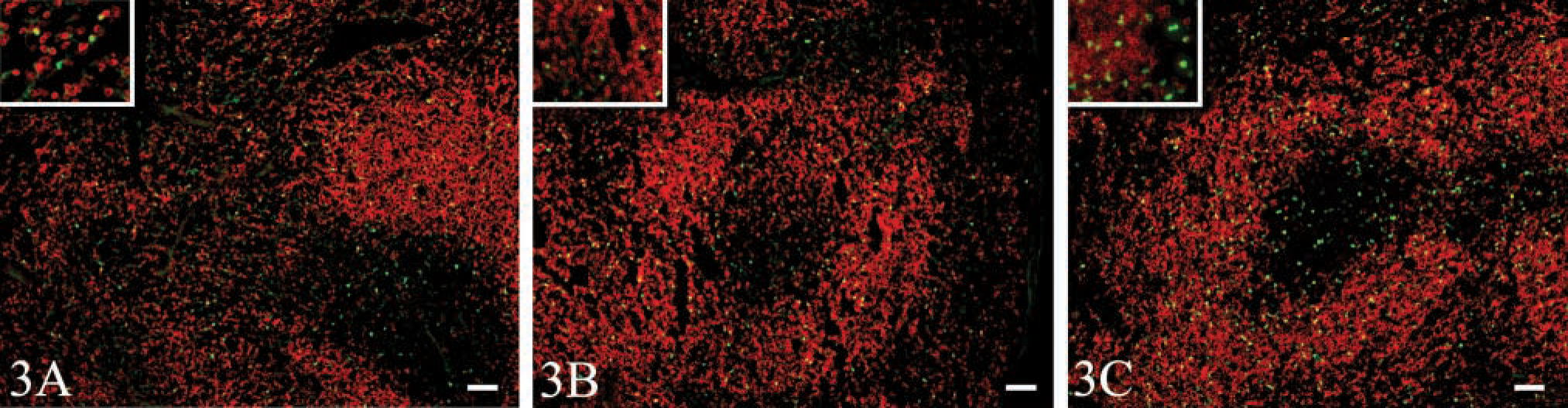
Perfusion-fixation maintains EGFP fluorescence but high perfusion pressures disrupt splenic architecture. EGFP-transgenic splenocytes were adoptively transferred to nontransgenic recipients. After 2 days, recipient mice were sacrificed and the thoracic cavity was opened to allow manipulation of the heart. Mice were perfused through the right ventricle with 20 ml of 2 U/ml heparin in PBS, followed by 20 ml of 4% formalin and 7% picric acid in PBS, followed by 20 ml of 10% sucrose in PBS at a flow rate of (
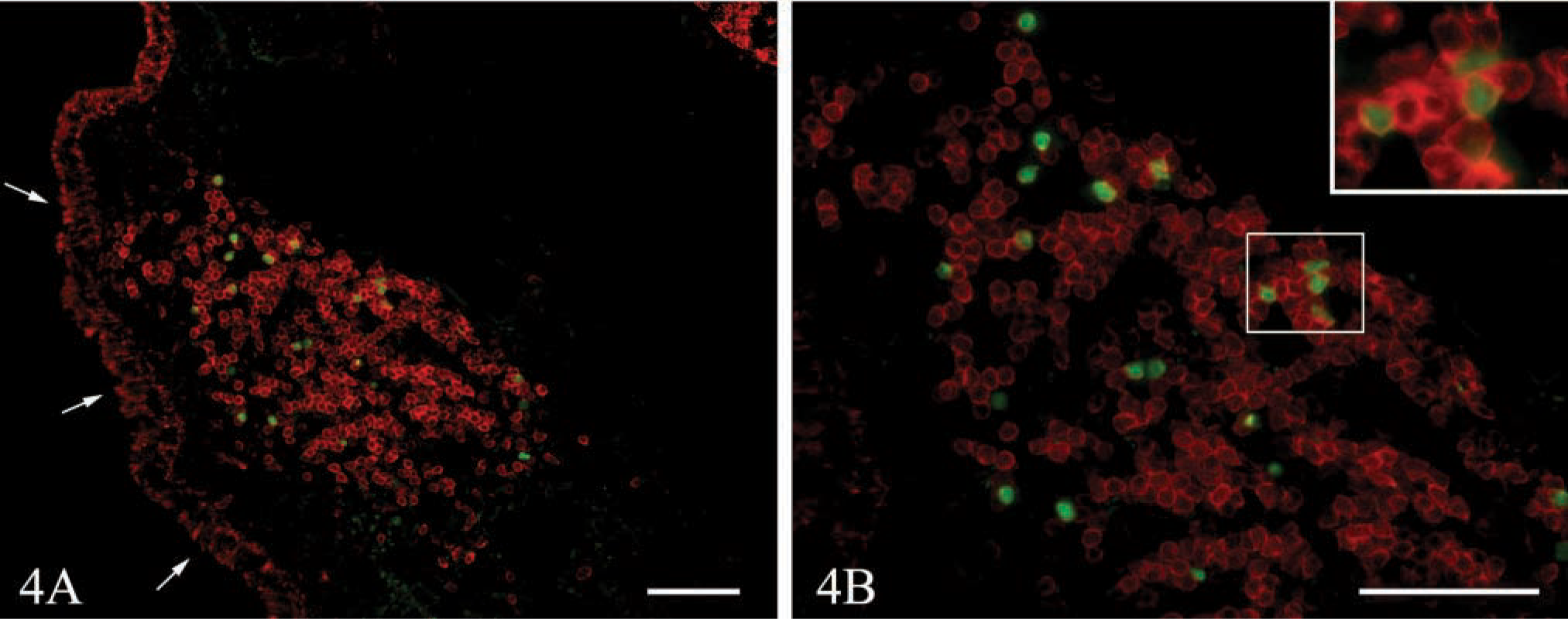
The fluorescence of EGFP-expressing cells is maintained after perfusion-fixation and decalcification of NALT. EGFP-transgenic splenocytes were adoptively transferred to nontransgenic recipients. After 2 days, recipient mice were sacrificed and perfused by gravity drip through the right ventricle with 20 ml of 2 U/ml heparin in PBS, followed by 20 ml of 4% formalin and 7% picric acid in PBS, followed by 20 ml of 10% sucrose in PBS. Mice that had been fixed by perfusion were decapitated and the skin, lower jaw, and incisors of the upper jaw were removed. The remaining tissue was decalcified with 7% EDTA and 10% sucrose in PBS over several days. Decalcified heads were embedded in OCT and snap-frozen over liquid nitrogen. Tissues were sectioned (7 μm), and probed with anti-B220 (red). Images were obtained at X20 original magnification (
Discussion
Although the visualization of EGFP in living cells usually poses few problems (Baumann et al. 1998; Endow 2001), the detection of EGFP in tissue sections can be more difficult because soluble EGFP molecules can be lost if cell integrity is disrupted by freezing, sectioning, or permeablization. Furthermore, the fluorescence of EGFP is dependent on its conformation (Brejc et al. 1997). Therefore, fixation protocols that immobilize EGFP may also destroy its usefulness as a fluorescent reporter.
Other reports have demonstrated that the fluorescence of EGFP is maintained in frozen sections of fixed tissues (Shariatmadari et al. 2001), but this is the first report to systematically compare multiple methods of fixation to determine which method optimally preserved EGFP fluorescence. Although the immersion of tissues in 4% paraformaldehyde and subsequent freezing have been used to preserve the fluorescence of EGFP-expressing cells in thick sections (Shariatmadari et al. 2001), we found that this technique did not maintain the brightness of EGFP in thin sections as well as the combination of 4% formalin and 7% picric acid in PBS (Table 2). Although some investigators have stated that EGFP could be detected in frozen sections without fixation (Manfra et al. 2001; Sata et al. 2002), this was not our experience. Furthermore, even the addition of sucrose as a cryopreservative did not maintain EGFP fluorescence in the absence of fixation (Figure 1C).
The requirement for a fixative suggested that soluble EGFP was simply leaking out of the sectioned cells. This hypothesis was supported by data showing that EGFP could not be detected by anti-GFP antibodies in unfixed frozen sections (Figure 1M-1O). However, our attempts to immobilize EGFP after sectioning by fixing sections with acetone, formalin, or paraformaldehyde were uniformly unsuccessful (not shown), suggesting that the loss of soluble EGFP occurs rapidly after thawing. The loss of EGFP is particularly acute in our experimental system, in which cells from mice transgenic for a soluble form of EGFP are adoptively transferred to nontransgenic recipients. In other systems, in which EGFP is fused to protein domains that are anchored to the cytoskeleton, have transmembrane regions, or are otherwise immobilized in the cell, the loss of EGFP may not be as severe.
Although the loss of EGFP signal in our experimental system is clearly related to loss of EGFP protein rather than to a change in the conformation of EGFP, some procedures do appear detrimental to EGFP conformation and fluorescence. For example, we have found that EGFP fluorescence decreases with extended periods of fixation (not shown). Furthermore, other investigators have demonstrated that the fluorescence of EGFP is lost on paraffin embedding of paraformaldehyde-fixed tissues (Leithauser et al. 2001). Therefore, even though we have found that a wide variety of fixation conditions retain EGFP protein and preserve EGFP fluorescence, not all fixation procedures are compatible with EGFP fluorescence.
Although multiple methods of fixation were able to preserve the fluorescence of EGFP, the fixation process often resulted in cells with a desiccated appearance and gaps between cells. This problem was partially overcome by the addition of 10% sucrose to the fixative. With the addition of sucrose, cells retained their rounded shapes, even though some gaps remained between the cells. In addition, although the perfusion-fixation method was best suited for uniform fixation of tissues that were not easily immersed, such as the NALT, perfusion often resulted in disrupted splenic architecture, particularly at high rates of perfusion (Figure 3). This is due to the direct connection of the spleen to the blood. The architecture of other lymphoid organs, such as the NALT, lymph nodes, and Peyer's patches, were less affected by perfusion pressure because they are continuous with lymphatics rather than with blood vessels.
Fixation of lymphoid organs under conditions that best preserve the fluorescence of EGFP also maintains the ability of multiple antibodies to be used for immunofluorescence (Figure 2). These antibodies are used to define the architecture of lymphoid organs, which consists of separated B- and T-cell areas and their associated dendritic cells. Therefore, the placement of EGFP-expressing cells in lymphoid organs can be easily determined using these methods.
In summary, we describe a method of preparing murine lymphoid tissues that optimally immobilizes EGFP and preserves its fluorescence, while simultaneously allowing the concurrent visualization of multiple cell surface markers and maintaining the cell architecture of the spleen and other lymphoid organs. Although we have demonstrated the utility of this method to detect adoptively transferred EGFP-expressing cells, similar methods could also be used to detect EGFP produced under the control of gene-specific or tissue-specific promoters.
Footnotes
Acknowledgements
Supported by the Trudeau Institute and by NIH grant AI43589 (to T.D.R.).
We would like to thank Dr Frances Lund for critically reviewing this manuscript.