
Abstract
Angiogenesis is implicated in a variety of human pathologies and may also play a role in the progression of heart failure. We have studied the expression of members of the vascular endothelial growth factor (VEGF) and the angiopoietin families and their receptors in mice lacking the mitochondrial transcription factor A. These mice lack functional respiratory chain activity in their myocytes and develop dilated cardiomyopathy (DCM) postnatally. We studied the hearts of the knockout mice by in situ hybridization, Western blotting analysis, and immunohistochemistry. VEGF-A mRNA and protein levels were elevated in the myocardium of the knockouts. Levels of the hypoxia inducible transcription factor 1 alpha (HIF1α) and of glyceraldehyde-3-phosphate dehydrogenase transcripts were also increased, whereas those of angiopoietin−1 and −2 were reduced. Despite the striking upregulation of VEGF-A, there was no increase in capillary density in the knockout hearts. This study suggests that a disturbance in angiogenesis may contribute to the pathogenesis of DCM.
Dilated cardiomyopathy (DCM) is a disease of the heart muscle characterized by systolic dysfunction and ventricular dilatation. Heart failure and cardiac arrhythmias develop and are the major causes of death in DCM patients. The etiology of generalized DCM is diverse and includes mutations in sarcomeric genes or mitochondrial DNA, viral infections, toxins, and endocrine disorders (Richardson et al. 1996). Coronary artery disease causes local cardiac damage and can give rise to ischemic cardiomyopathy (ICM), which is one of the most common causes of heart failure in the Western world. How these diverse factors lead to a mutual disease phenotype and which mechanisms contribute to the deterioration of heart function in DCM are still largely unknown.
Angiogenesis is implicated in a variety of human pathologies such as cancer, ICM, and retinopathy (Ferrara 1999). A multitude of factors control the angiogenic response, but two families of proteins have more endothelial specific effects: the vascular endothelial growth factor (VEGF) and the angiopoietin families (Bussolino et al. 1997). Of the five VEGF genes known in humans, VEGF-A is the major angiogenic factor. Acting through the VEGF receptor 2 (VEGFR2), VEGF-A is involved in all steps leading to the development of immature vessels (Carmeliet and Collen 1999). VEGF-A is of vital importance for blood vessel formation both in the adult and during embryogenesis, and loss of a single allele is embryonically lethal (Ferrara et al. 1996). The angiopoietins regulate the responsiveness of the endothelial cells to VEGF-A and are important not only for vessel growth but also for vessel stabilization and regression (Suri et al. 1996; Davis and Yancopoulos 1999; Partanen and Dumont 1999). If the two major isoforms of VEGF-A (VEGF-A-164 and VEGF-A-188) are specifically knocked out in the murine heart, leaving only VEGF-A-120, which constitutes 5% of total cardiac VEGF-A, the mice develop DCM and die shortly after birth (Carmeliet et al. 1999). In addition, if collagen2a1-producing tissues lose VEGF-A (via a cre-loxP conditional knockout), mice that survive the embryonic period succumb to DCM (Haigh et al. 2000). These studies suggest that a disturbance in angiogenesis may contribute to the progression of DCM. This hypothesis is supported by a study of 21 transplant patients with DCM who had decreased cardiac levels of VEGF-A and VEGFR1 mRNA as well as a lower capillary density than in controls (Abraham et al. 2000).
Mitochondrial dysfunction may be both a cause and a consequence of DCM. Mutations in mtDNA can lead to DCM, often as part of a multiorgan syndrome (DiMauro and Hirano 1998). A deficiency in reactive oxygen species (ROS) scavengers (such as manganese superoxide dismutase) which protect sub-units in the mitochondrial respiratory chain from free radical damage, also results in DCM (Li et al. 1995). Conversely, some cases of idiopathic DCM exhibit decreased oxidative phosphorylation and/or mtDNA mutations as secondary phenomena (Arbustini et al. 1998; Jarreta et al. 2000; Quigley et al. 2000). In addition, mitochondria may be involved in the upregulation of angiogenesis via their ability to sense oxygen levels (Chandel and Schumacker 2000). Therefore, we chose to examine the expression of the VEGF and angiopoietin family members and their receptors in a unique mouse model of DCM caused by mitochondrial respiratory chain deficiency (Wang et al. 1999). These mice have a heart-specific knockout of the mitochondrial transcription factor A (Tfam) and therefore cannot perform oxidative phosphorylation. We found an increase in VEGF-A mRNA and protein levels using in situ hybridization and Western blotting analysis. However, surprisingly, VEGF-A did not elicit an angiogenic response.
Materials and Methods
Tissue Preparation
Hearts from nine (TfamloxP/TfamloxP + Ckmm-cre) mice at a terminal stage of heart failure (3 weeks of age) and from their healthy (TfamloxP/TfamloxP) littermates were snap-frozen in liquid nitrogen directly after sacrifice (Wang et al. 1999). The hearts were stored at −70C before protein extraction or cryosectioning of hearts embedded in Tissue-Tek OCT compound (Miles; Elkhart, IN). The Lab Animal Ethical Committee of Northern Stockholm has approved this study.
In Situ Hybridization
Oligonucleotide ISH was performed as previously described (Lagercrantz et al. 1996; Lidman et al. 1999) on mouse hearts at a terminal stage of heart failure. Briefly, 14-μm cryostat cross-sections (Leica CM3000; Wetzlar, Germany) were thawed onto Superfrost Plus slides (Menzel-Glaser; Braunschweig, Germany) and stored in sealed black boxes at −20C until used. The sections were cut from the middle third of the mouse hearts and contained the open lumens of both the left and right ventricles. Littermate mutant and control hearts were placed on the same slide to exclude slide-to-slide variation in the analysis of the signal intensity. The oligonucleotide probes (Genset; Paris, France) were designed to have a 55% GC content using the oligo 4.0 program (National Biosciences; Plymouth, MI) and were genespecific on a BLAST search. To test the specificity of the probes, they were first hybridized to a panel of normal mouse tissues. Two oligomers were used separately for each mRNA (Table 1) and each pair displayed highly similar expression patterns that corresponded to previously published data. In contrast, non-overlapping expression patterns were evident when the hybridization signals of the different probe pairs were compared.
The probes were labeled at the 3′ end with [35S]-deoxyadenosine-α[thio]triphosphate (NEN; Boston, MA) using terminal deoxynucleotidyl transferase (FPLC pure; Amersham Pharmacia Biotech, Uppsala, Sweden) to a specific activity of 1.6–4 × 109 cpm/μg. Unincorporated labeled nucleotide was removed with QIAquick Nucleotide Removal Kit (Quiagen; Hilden, Germany). Hybridization was performed at 42C for 16–18 hr on slides pretreated with 0.005% acetic anhydride in acetone. The hybridization mixture contained 50% formamide (GT Baker Chemicals; Amsterdam, The Netherlands), 4 × SSC (1 × SSC = 0.15 M NaCl and 0.015 M sodium citrate), 1 × Denhardt's solution [0.02% each of polyvinyl-pyrrolidone, bovine serum albumin (BSA), and Ficoll], 1% sarcosyl (N-lauroylsarcosine; Sigma, St Louis, MO), 0.02 M phosphate buffer (pH 7.0), 10% dextran sulfate (Amersham Pharmacia Biotech), 250 μg/ml yeast tRNA (Sigma), 500 μg/ml sheared and heat-denatured salmon sperm DNA (Sigma), and 200 mM dithiothreitol (DTT; Amersham Pharmacia Biotech). After hybridization the slides were washed in 1 × SSC at 60C, dehydrated in ethanol, and exposed to autoradiographic film (Hyperfilm Beta-max; Amersham Pharmacia Biotech) followed by dipping in NTB 2 nuclear track emulsion (Kodak; Rochester, NY). After exposure for 1 day (18srRNA), 5 days (GAPDH, COX, ANF), 4 weeks (VEGF-A), or 5 weeks (all the others), the sections were developed in D-19 developer (Kodak-Pathé; Chalon-Sur-Saone, France) for 4 min, fixed in Unifix (Kodak-Pathé), and mounted. The digitalized darkfield image of the dipped slides of four pairs of hearts was recorded by a Kappa video camera connected to a Leica DM RBE microscope (Mikroskop System; Näsviken, Sweden) and the silver grain density was measured using the NIH Image 1.55VDM program (NIH; Bethesda, MD). The pixel intensity measurements of the silver grains in the tissue have previously been demonstrated to correlate well to the actual number of silver grains (Piehl et al. 1995). A minimum of five randomly selected fields with an average area of 0.3 mm2 was measured in each heart. The ratio of the signal in the knockouts compared to that in their healthy littermate control (measured from the same slide under the same conditions) was calculated for each pair. For VEGF-A, the grain density was also measured within an area of 1.72 × 10-4 mm2, corresponding approximately to one cell. A total of 50 measurements (cells) were made for each heart. The in situ slides were photographed in darkfield for optimal visualization of the in situ signal, in brightfield, and also with differential interference contrast (DIC) to reveal structural details in the tissue. A digital camera (Axiocam; Carl Zeiss, Jena, Germany) connected to a Zeiss Axioscope 2 MOT microscope was used.
Oligomers used for ISH

Western Blotting Analysis
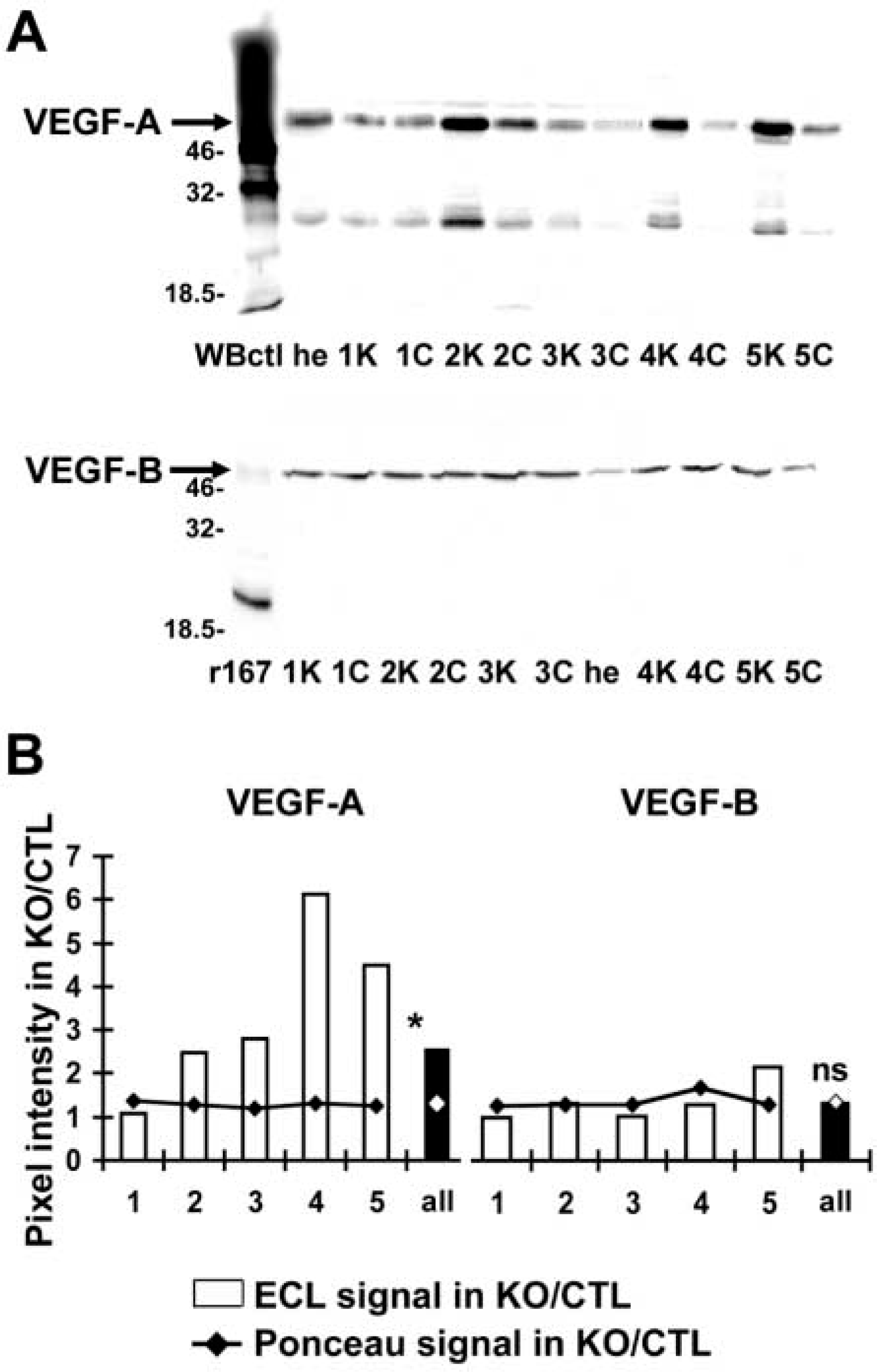
Protein was extracted from the hearts of five terminal stage knockout mice and their littermate controls as well as from unrelated normal mouse heart according to standard procedures. A total of 30 μg of protein and 3 μl kaleidoscope protein size standards (BioRad Laboratories; Hercules, CA) in 1 × SDS buffer containing 50 mM DTT was heated to 85C for 5 min and then electrophoresed on 12% SDS-PAGE gels in a mini-Protean 2 electrophoresis system (BioRad). Proteins were blotted onto 0.45-μm nitrocellulose membranes (Protran; Schleicher & Schuell, Dassel, Germany), stained with Ponceau S, and scanned into the computer to use as a loading control. After blocking in 4% non-fat milk, the membranes were incubated with anti-VEGF-A (sc-7269 or sc-152) 0.4 μg/ml (Santa Cruz Biotechnology; Santa Cruz, CA) or with anti-VEGF-B (AF751) 0.2 μg/ml (R&D Systems; Oxford, UK) at 4C overnight. The membranes were then incubated with secondary horseradish peroxidase-conjugated antibodies at 0.2 μg/ml dilution (Santa Cruz Biotechnology). Detection was carried out by Enhanced ChemiLuminescence (Amersham Pharmacia Biotech) and the proteins were visualized in a CCD camera using the luminescent image analyzer LAS-1000plus (Fujifilm; Tokyo, Japan). The immunostaining pattern of the two VEGF-A antibodies was identical. The ECL signal of antibody sc-7269 and the intensity of the scanned image of the Ponceau S stain (loading control) were quantified using the Image Gauge V3.3 program (Fujifilm). The ratio of the immuno- or Ponceau S staining signal in the knockouts relative to the controls was calculated for each of the five littermate pairs. Recombinant VEGF-B-167 and VEGF-B-186 were produced in COS cells (unpublished data) and used as positive controls, as was a 46-kD fusion protein of VEGF-A (Santa Cruz Biotechnology).
Immunohistochemistry
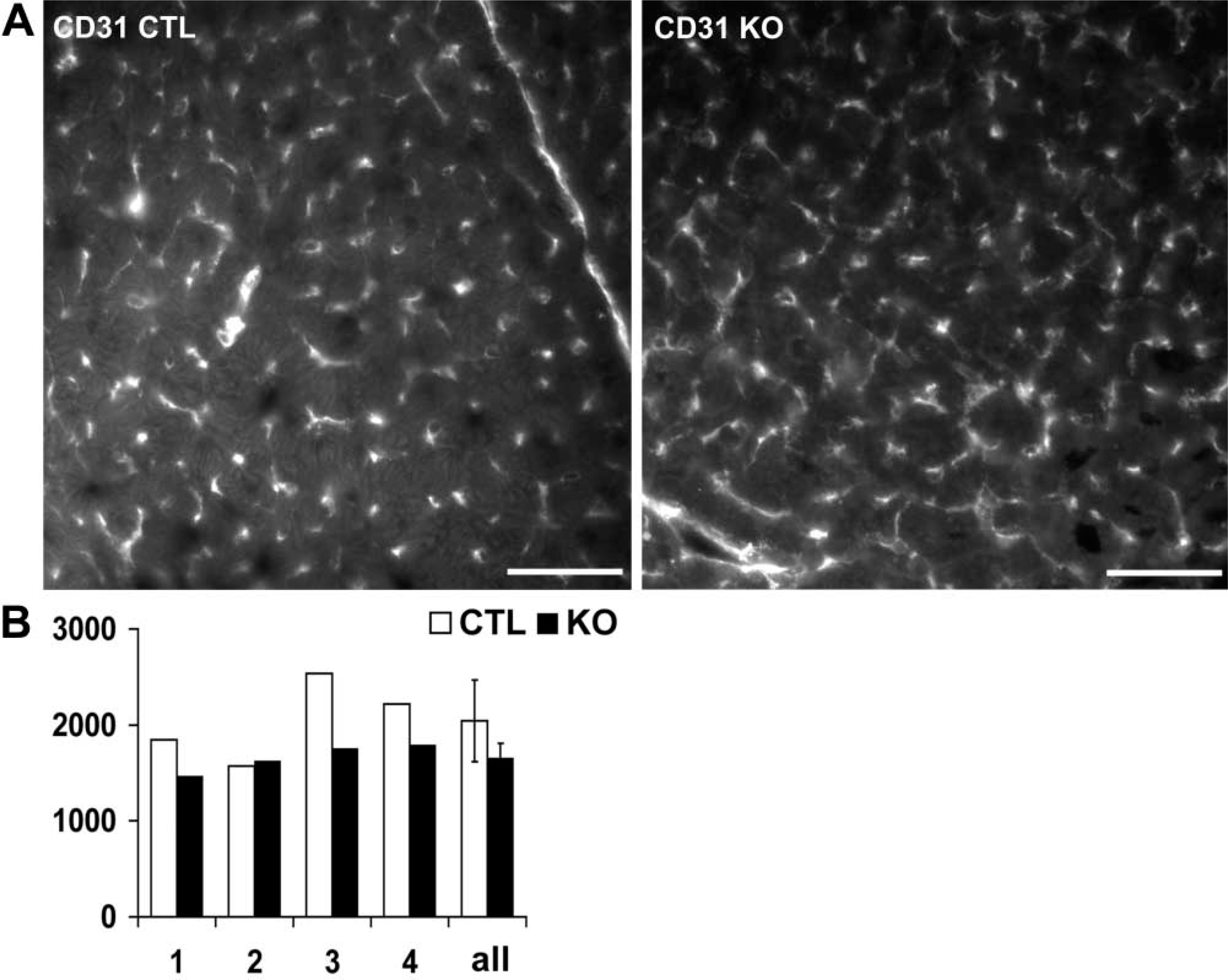
For blood vessel analysis, immunohistochemistry (IHC) was performed on the same hearts as the ISH experiments. The sections were cut from the third of the heart closest to the apex, with the left ventricle lumen still visible. For the blood vessel staining, 8-μm cryostat cross-sections were thawed onto Superfrost Plus slides (Menzel-Glaser) and stored at −20C until used. The tissues were fixed in 100% ice-cold acetone for 10 min followed by 4% paraformaldehyde for 1 min. After blocking in 3% donkey serum for 30 min, the slides were incubated with primary antibodies in PBS-Triton 0.1% buffer overnight at 4C, followed by Cy3-conjugated secondary antibodies at 2.4 μg/ml (Jackson Immuno-Research Lab; West Grove, PA) for 1 hr at room temperature. Antibodies used were rat anti-mouse CD31/PECAM (01951A from PharMingen; San Diego, CA) at 0.5 μg/ml or rabbit anti-mouse von Willebrand Factor (A0082 from Dako; Glostrup, Denmark) at 16 μg/ml. As negative controls, we omitted the primary antibody or used normal rat/rabbit serum (Dako) at a concentration of 10 μg/ml. The image of the fluorescent blood vessels was recorded by a digital camera (Axiocam; Carl Zeiss) connected to a Zeiss Axioscope 2 MOT microscope. Blood vessels were counted in four pairs of hearts in a total cross-sectional area of 0.56 mm2 per heart. All free-standing positive signals that were not visible branches of another vessel were counted. Vessels crossing the lower and left borders of the field were not counted.
For the VEGF-A staining, 10-μm sections were cut, air-dried for 5–10 min, and then fixed in 100% ice-cold acetone for 10 min. The slides were blocked with 0.3% H2O2, then with 10% donkey serum in PBS-0.1% Tween, followed by blocking endogenous avidin and biotin (avidin-biotin blocking kit SP-2001 from Vector Laboratories; Burlingame, CA). Slides were incubated with rabbit-anti-VEGF-A (sc-152 from Santa Cruz Biotechnology) at 0.2 μg/ml or with normal rabbit serum at 0.4 μg/ml overnight at 4C. After incubation with a donkey anti-rabbit biotinylated secondary antibody (Jackson ImmunoResearch Lab), the slides were incubated with ABC Elite (Vector Laboratories). Antibody detection was performed using the peroxidase substrate diaminobenzidine tetrahydrochloride (DAB) (SK-4100; Vector Laboratories). The sections were counterstained with hematoxylin (Apoteket; Malmö, Sweden).
Statistics
All data are presented as the mean ± SD. Statistical analyses were carried out using GraphPad Prism 3.0 (GraphPad Software; San Diego, CA). The ISH values and the blood vessel counts were analyzed with the paired Student's t-test. The values of the Western blots of VEGF-A and VEGF-B were analyzed using the Wilcoxon signed rank test on the ratios of the knockout to the controls. The results were considered significant at a value of p<0.05.
Results
Further Characterization of the Mouse Model
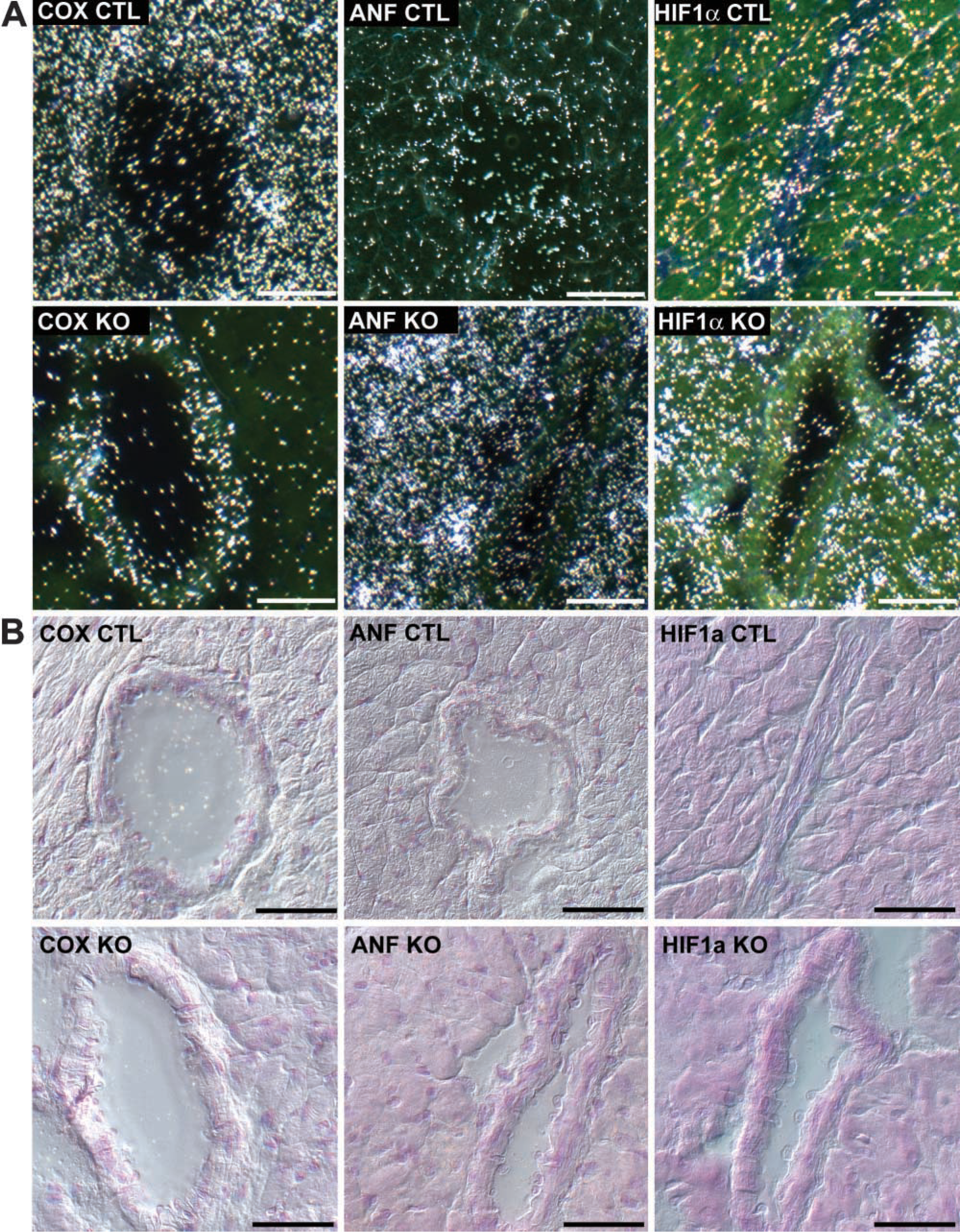
The knockout mice at a terminal stage of heart failure have been well characterized regarding morphology, histology, and function in previous publications (Wang et al. 1999; Wang et al. 2001). ISH of the mRNA of a mitochondrial cytochrome-c oxidase subunit (COX) confirmed earlier Northern blotting results and clearly demonstrated the cardiomyocyte-specific lack of mitochondrial mRNA due to knockout of the transcription factor Tfam (Figures 1 and 5). Atrial natriuretic factor (ANF), a marker of heart failure, was increased in the myocytes. In addition, the mRNA signal of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Wang et al. 2001) and the glucose transporter type 1 (Glut-1) were both elevated, indicating that the hearts deficient in oxidative phosphorylation had a compensatory increase in glucose metabolism (Figure 1). The robust 18srRNA signal remained constant in all samples (Figure 1).
Expression Analysis of VEGF-A and VEGF-B
The most striking alteration among the angiogenesis factors (Figure 1) was the increase in transcript levels of VEGF-A. ISH demonstrated a significant increase in VEGF-A mRNA by 1.7-fold in the DCM hearts of this model. The hybridization signal for VEGF-A displayed a punctate pattern with dense grain accumulations over cells identified as cardiomyocytes by histopathological analysis (Figure 2). However, as previously described (Marti and Risau 1998), VEGF-A was expressed only by a subset of normal (and Tfam-deficient) cardiomyocytes. When measured at higher magnification over single positive cells (50 in each heart), the VEGF-A mRNA levels were located centrally in the cell (Sköld et al. 2000) and displayed a 3.4-fold increase in the positive cardiomyocytes of the knockouts compared to their healthy controls. This punctate expression pattern contrasted to the uniform pattern of the VEGF-B mRNA, which was not altered in the DCM hearts (Figures 1 and 2).
Western blotting analysis confirmed that VEGF-A was elevated at the protein level. Quantification of the Western blotting signal demonstrated an augmentation of VEGF-A protein in four of the five knockout hearts tested. In contrast, the protein levels of VEGF-B remained constant. Only the 167 amino acid form of VEGF-B was expressed in these mouse hearts (Figure 3).
We performed IHC using an antibody against VEGF-A (A-20) that has previously been used successfully in rat tissues (Ogunshola et al. 2000; Tomanek et al. 2001). The staining pattern was indistinguishable between the knockouts and the controls, suggesting that there was no functional difference of VEGF-A between the animals. The VEGF-A antibody stained the cytoplasm of the cardiomyocytes and also the endothelial cells of the blood vessels, indicating that VEGF-A protein was secreted from the cardiomyocytes and was functional (Figure 4).
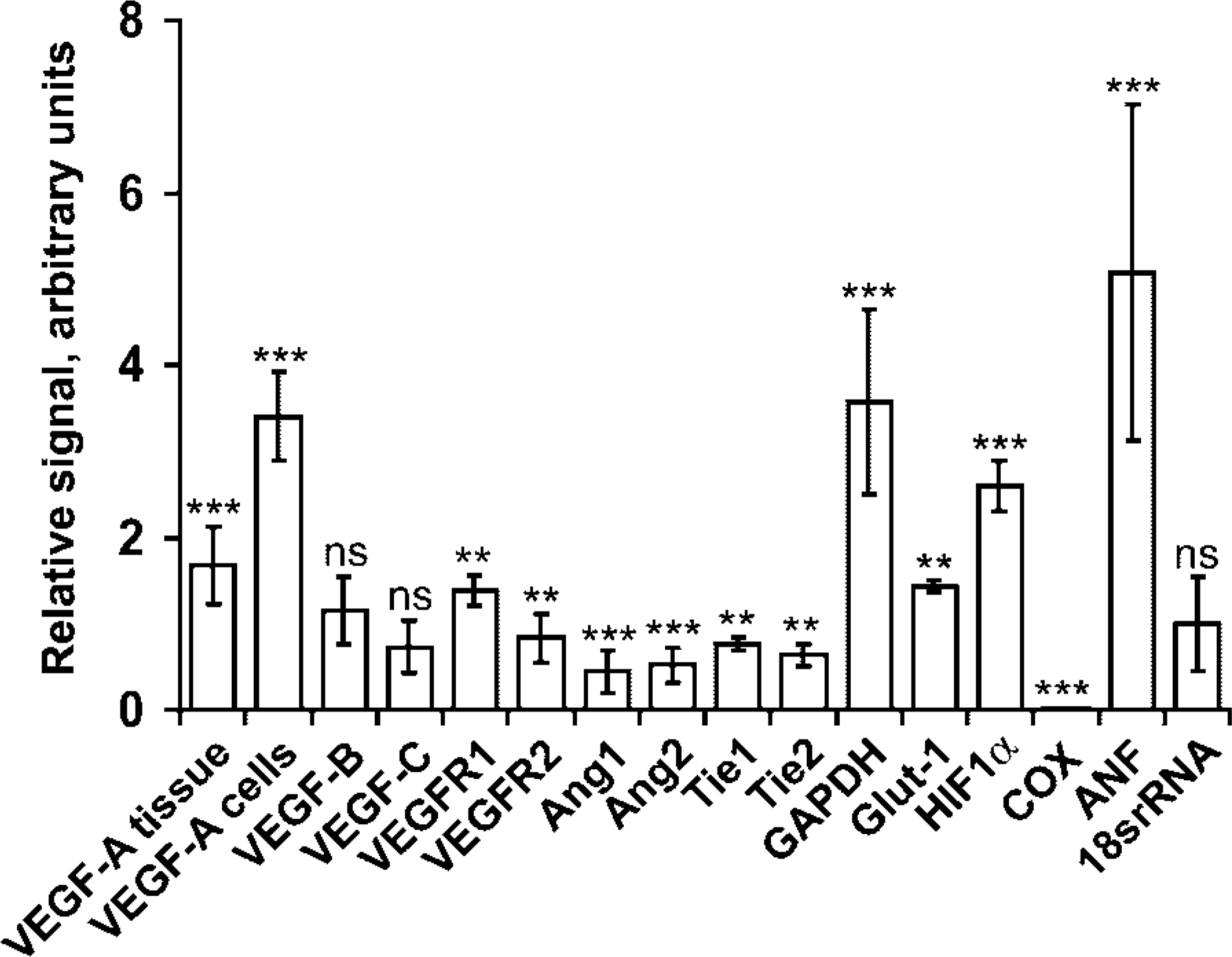
Measurements of RNA ISH signals of all factors studied. Results are presented as a ratio of the average signal of the knockout mice compared to their littermate controls, with the controls set to 1. The SD of the average value is indicated. Differences were calculated using Student's paired t-test and considered significant if p<0.05 (∗), p<0.01 (∗∗), or p<0.001 (∗∗∗). The signals were measured in fields of on average 0.3 mm2 except for VEGF-A cells, for which the signal was measured within an area of 1.72 × 10-4 mm2, corresponding approximately to one cell.
ISH of Other Angiogenesis Factors
The transcript levels of the main receptor involved in angiogenesis, VEGFR2, were constant, whereas VEGFR1 was subtly (1.4-fold) elevated in the knockout hearts. VEGF-C had unaltered mRNA signals. Both angiopoietins, their receptor Tie-2, and the orphan receptor Tie-1 displayed a weak mRNA signal over the blood vessels, with a slight reduction in the DCM hearts (Figure 1). Because hypoxia is a powerful inducer of angiogenesis and may involve mitochondrial function, we also analyzed the mRNA of the hypoxia-inducible transcription factor 1 alpha (HIF1α), which was upregulated by 2.6-fold in the cardiomyocytes (Figures 1 and 5).
Blood Vessel Density
Because the levels of VEGF-A were elevated, we investigated whether there were any signs of neovascularization in the DCM hearts. Blood vessels (large vessels as well as capillaries) were identified with immunofluorescence staining for CD31/PECAM, a membrane protein expressed constitutively on endothelial cells (DeLisser et al. 1994; Scholz and Schaper 1997), and confirmed using an antibody against vWF (data not shown). No signal was detected when the primary antibody was omitted or substituted for normal serum. Blood vessels were counted from the digitalized image of representative cross-sections in a total of eight mouse hearts. The density of blood vessels in the knockouts demonstrated no increase compared to their littermate controls (Figure 6).

RNA ISH photographs of VEGF-A and VEGF-B. VEGF-A in control (CTL) and knockout (KO) mice (from the same slide) is shown at x 75 magnification in darkfield photographs and at x 300 magnification in brightfield photographs. VEGF-B in control (CTL) and knockout (KO) hearts (from the same slide) is shown at x75 magnification in darkfield photos of the same area as VEGF-A. Note the punctate pattern of VEGF-A expression, which contrasts to the more general expression of VEGF-B. The VEGF-A signal is clearly elevated in the knockout hearts in a subset of the cardiomyocytes.
Discussion
In this study we have demonstrated that the expression levels of several of the major factors involved in angiogenesis are affected in DCM of mitochondrial etiology. We examined the expression of the members of the VEGF and angiopoietin families and their receptors by ISH in a mouse model of dilated DCM caused by mitochondrial dysfunction (Wang et al. 1999). These mice have a cre-loxP conditional knockout of Tfam, a nucleus-encoded factor that is required for mtDNA transcription. Cre-recombinase, under the control of the creatinine kinase promotor, causes postnatal excision of the lox-P flanked Tfam alleles specifically in myocytes. A large proportion of the cardiomyocytes lack Tfam and therefore cannot transcribe mtDNA-encoded respiratory chain subunits (including COX), causing severe oxidative phosphorylation deficiency. The cells become hypertrophic and accumulate swollen, abnormal mitochondria. The mRNA levels of ANF, a marker of cardiac failure, increase in the ventricles. In addition, the ATP-deficient cardiomyocytes stimulate glycolysis (as demonstrated by the increase in GAPDH). However, there is no fibrosis or infiltration of inflammatory cells (Wang et al. 2001). DCM develops and progresses to manifest heart failure with atrioventricular conduction blocks, leading to death by the age of 2–4 weeks.
Our results diverge from those of Abraham et al. (2000) in their study on the expression of VEGF-A, -B, and -C and their receptors in human transplant patients with cardiomyopathy mentioned above. Their study showed a correlation between levels of VEGF-A and capillary density. Patients with idiopathic DCM had 50% lower VEGF-A165, VEGFR1, and capillary density (with constant VEGFR2 and VEGF-B levels and increased VEGF-C). Conversely, patients with ICM had elevated VEGF-A (and VEGF-C) levels and a higher capillary density compared to controls. However, we clearly demonstrate an increase in VEGF-A in mitochondrial DCM without a concomitant increase in capillary density. The discrepancy between the levels of cardiomyocyte VEGF-A in our mice and in the patients of Abraham et al. (2000) could reflect etiological differences between the two groups of DCM. Mitochondrial dysfunction may trigger a distinct pathway that leads to elevation rather than a decrease in VEGF-A.
Why does the 3.4-fold increase of VEGF-A fail to elicit an angiogenic response in our mouse model? Angiogenesis is a tightly regulated process and VEGF-A is, for example, only angiogenic in a permissive environment. Exogenous VEGF-A injected into chick embryos failed to induce vascularization of the normally avascular cornea and retina, despite expression of VEGF-A and striking vessel growth in the surrounding tissues (Schmidt and Flamme 1998). This indicates that a correct balance of VEGF-A and auxiliary factors in relation to inhibitors is necessary for angiogenesis to be possible. The fact that the major VEGF-A receptor, VEGFR2, is not upregulated could, in theory, explain the lack of a response to VEGF-A. However, this is unlikely, as VEGFR2 remains constant even when there is active angiogenesis (Abraham et al. 2000).
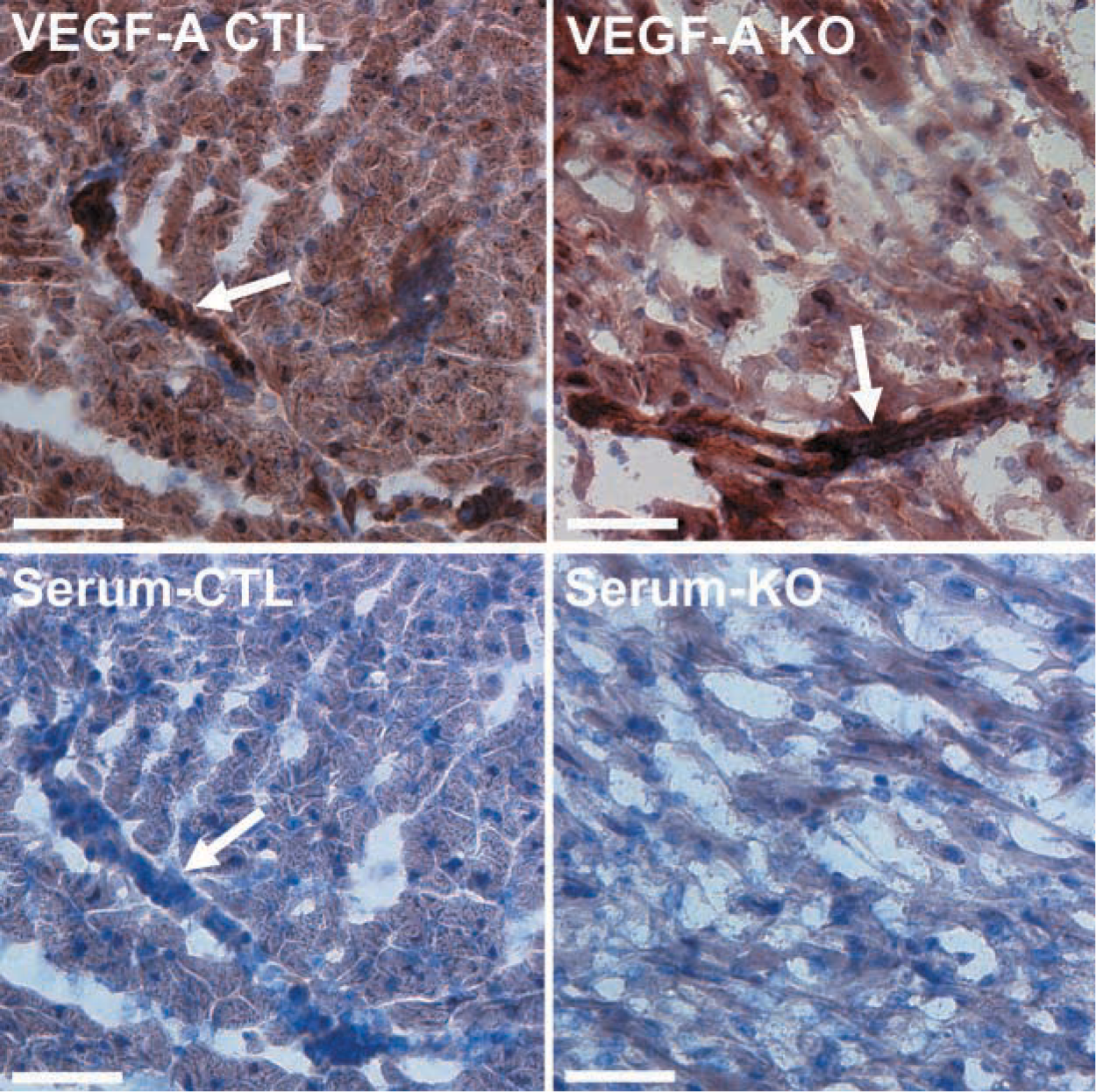
Immunohistochemical detection of VEGF-A (top) and negative control serum (below) in terminal stage DCM mouse hearts. There is diffuse cytoplasmic staining in the cardiomyocytes and a strong signal in the blood vessels (arrows) of both the control (CTL) and the knockout (KO) animals, indicating that VEGF-A is secreted and functional. Bars = 50 μm.
The presence of leukocytes may determine whether a vascular response will ensue. However, inflammation is not a prerequisite for angiogenesis (Schmidt and Flamme 1998). For example, ovarian angiogenesis occurs without inflammation, whereas tumor angiogenesis is associated with prominent leukocyte infiltration (Goede et al. 1999). It has yet to be determined whether the absence of immune cell infiltration in our model of cardiac failure contributes to the lack of angiogenesis.
Many other factors may be involved in the regulation of neovascularization. One such factor is the presence or absence of auxiliary proteins, such as the angiopoietins. The angiopoietins act synergistically with VEGF-A (Asahara et al. 1998) and play a role in both angiogenesis and cardiac remodeling (Suri et al. 1996; Maisonpierre et al. 1997). In our model, the expression of both angiopoietins and their receptor was reduced, which may have contributed to the lack of angiogenesis in these mouse hearts.
Angiogenesis has been most extensively studied in ischemic settings. Hypoxia is a powerful inducer of angiogenesis via the action of HIF1α (Liu et al. 1995; Forsythe et al. 1996). HIF1α mRNA was elevated in our DCM model and has previously been found to rise parallel to progression of heart failure in CHF146 cardiomyopathic hamsters and in post-infarction heart failure in rats (Kakinuma et al. 2001). Although HIF1α is also regulated at the protein level, the fact that four of its target genes were stimulated (VEGF-A, VEGFR1, GAPDH, and Glut-1) (Ebert et al. 1995; Liu et al. 1995; Gerber et al. 1997; Graven et al. 1999) indirectly suggests that the elevated HIF1α mRNA levels in our model correlate with increased HIF1α activity.
In summary, mice lacking mitochondrial function in their cardiomyocytes develop DCM. They have an increase in VEGF-A mRNA and protein (but not VEGF-B or VEGF-C) in their cardiomyocytes without a concomitant increase in capillary density. In addition, we found an elevation of HIF1α mRNA in the myocardium. In conclusion, upregulation of HIF1α and VEGF-A was not sufficient to induce neovascularization in this model. Therefore, the regulation of angiogenesis appears to be more complex than expected, and this may have implications for the use of angiogenic therapy in patients with, e.g., myocardial infarction.
Footnotes
Acknowledgements
Supported by grants from the Swedish Cancer Society, Axel and Signe Lagerman's Foundation, Sigurd and Elsa Golje's Memorial Fund, the Swedish Medical Society, and Förenade Liv Group Insurance company.
We are grateful to Prof Magnus Nordenskjöld for his support. We thank Dr Nils-Göran Larsson for valuable help and for providing the Tfam knockout mice. We also thank Dr Anders Oldfors (Sahlgrenska Hospital) for expert histo-pathological guidance.