
Abstract
Primary cilia are microtubule-based organelles protruding from the surface of mammalian cells, which function as hubs for transducing both biochemical signals (such as Hedgehog) and mechanical force. Defects in cilia are associated with a group of genetic disorders called ciliopathies that disrupt the normal development of multiple organs, including the craniofacial skeleton. One such example is Ellis–van Creveld (EvC) syndrome, a ciliopathy mainly caused by mutations in EVC (EvC ciliary complex subunit1), a key component of the primary cilium essential for the transduction of Hedgehog signaling. Patients with EvC syndrome exhibit a wide spectrum of clinical phenotypes related to multisystemic involvement, including oral defects such as malformations of the teeth, oral vestibule, and ectopic frenula. These distinctive oral defects play a crucial role in the initial diagnosis. The oral vestibule and associated frenula are formed from an embryonic structure, known as the vestibular lamina (VL), which forms closely associated with the dental lamina that forms the teeth. Here we reveal the developmental mechanisms underlying the oral defects in EvC syndrome using Evc knockout mice. Evc mutants exhibited defects in teeth, frenula, and the oral vestibule, mirroring the oral traits of patients with EvC syndrome. A defect in proliferation, and downregulation of Gli1 and Sostdc1 during initial outgrowth, led to a shortened VL, although postnatal differentiation and opening of the VL was normal. In some mutants, the VL branched and formed an ectopic tooth germ, which could be partially mimicked by the overexpression of Wnt signaling in the VL. Notably, we observed both upregulation and downregulation of Gli1 expression, which was time and tissue specific, suggesting dynamic dysregulation of the normal GLIActivator and GLIRepressor balance. These findings provide novel insights into the underlying etiology of EvC syndrome and other ciliopathies and emphasize that structures developing in close proximity can exhibit divergent responses to the same mutation.
Introduction
Ellis–van Creveld (EvC) syndrome is an autosomal recessive disorder reported across diverse populations, which is mainly caused by the mutations in EVC or EVC2 (Ruiz-Perez et al 2000; Galdzicka et al 2002). The patients are typically characterized by disproportionate dwarfism, shortened limbs and ribs, hypoplastic nails, congenital heart disease, and oral abnormalities (Sasalawad et al 2013).
Distinct oral features in patients with EvC syndrome are consistent and pathognomonic, especially the noticeable abnormalities in the oral vestibule, frenula, and teeth, which are crucial for the early diagnosis of the syndrome (Mostafa et al 2005; Sasalawad et al 2013). Vestibular dysplasia includes broad median frenula, accessory labio-gingival frenula, hypertrophic or short frenula (frenulum breve), and anterior labio-gingival adhesions from a reduced or obliterated mucobuccal sulcus (Sasalawad et al 2013; Nethan et al 2017). Comparable vestibular abnormalities are also found in Weyers acrofacial dysostosis, which is also linked to mutations in EVC/EVC2 (Roubicek and Spranger 1984; Ruiz-Perez et al., 2000). A diverse array of dental anomalies has been reported in EvC syndrome patients, such as tooth fusion, missing teeth, malformed teeth, premature exfoliation, supernumerary teeth, and taurodontism (Nakatomi 2009; Nethan et al 2017).
The oral vestibule and associated frenula develop from the vestibular lamina (VL), an embryonic structure that arises adjacent to the tooth-forming dental lamina (DL). The VL extends as an invaginating sheet that opens to create a gap between the lips and the teeth (Qiu et al 2020; Qiu and Tucker 2023). In early embryonic development, the VL and DL have been shown to share a common origin (Qiu et al 2020), and the VL has been shown to have the potential to form toothlike odontomas under specific pathological conditions (Hovorakova et al 2020).
The Evc mutant mouse, generated by deleting Evc exon 1 and inserting a lacZ reporter cassette, models EvC syndrome by replicating the loss-of-function mutations observed in patients and faithfully mimics their physical characteristics (Ruiz-Perez et al 2007). Previous studies showed Evc expression in the mouth and tooth-forming areas and in the developing skeleton from E11.5 to P0 in mice, including the maxillary, mandibular, lateral nasal processes, and whisker follicles (Ruiz-Perez et al 2007).
EVC is localized to primary cilia, where it forms a complex with EVC2 to mediate Hedgehog signaling, acting downstream of smoothened activation to process GLI into activator forms (Blair et al 2011; Dorn et al 2012). EVC and EVC2 are mutually dependent for proper localization to primary cilia, and mutations in either gene impair the cellular response to Hedgehog ligands (Caparros-Martin et al 2013). Interestingly, patients with mutations in GLI1 share many of the skeletal features of EvC syndrome patients (Palencia-Campos et al 2017). Evc mutants have been reported to show defects in molar development, with asymmetrical loss of responsiveness to Shh, leading to first molar microdontia, delayed differentiation, and root defects (Nakatomi et al 2013).
Dysfunctions of the primary cilia have been linked to a range of syndromic disorders, referred to as ciliopathies (Berbari et al 2009). In addition to EvC syndrome, a diverse array of craniofacial defects including abnormal frenula have been observed in oral-facial-digital syndrome, which represents a distinct subgroup within the category of ciliopathies (Bruel et al 2017).
Previous studies on Evc mutants focused on dental defects (Ruiz-Perez et al 2007; Nakatomi et al 2013) but without analysis of the associated vestibule and frenulum defects, which are characteristic of EvC syndrome. Here we show that Evc mutants have shortened and branching VL and highlight the mechanisms underlying these defects. We reveal unexpected context-dependent effects on dental and oral tissues that will aid in our understanding of the complexity of phenotypes observed across ciliopathies.
Materials and Methods
Animals
The generation of Evc-/- mice has been previously described (Ruiz-Perez et al 2007). Evc mutants were bred on a mixed CD1/C57BL/6 background to enhance the vigor of the colony and were compared with their wild-type (WT) littermates. To label proliferating cells, BrdU (30 mg/kg, Life Technologies, 000103) was administered by intraperitoneal injection to Evc pregnant mice 2 h prior to culling. Male Gli1CreERT2; tdTomato mice were mated with female Evc heterozygous mice to generate Gli1CreERT2; tdTomato; Evc+/- mice, which were mated to Evc heterozygous females to generate Gli1CreERT2; tdTomato; Evc-/-. For overexpression of the canonical Wnt pathway in the VL, Sox2CreERT2 males were mated to β-catenin GOF female mice (Ctnnb1-lox(ex3)). To activate Cre in both lines, pregnant mice were injected with 75 mg/kg tamoxifen (20 mg/mL, dissolved in corn oil: approximately 200 µL injected per pregnant mouse) for 2 to 3 d before culling. The day of plug detection was marked as E0.5. All animal procedures were conducted following the guidelines set forth by King’s College London and the UK Home Office, with mice culled using Schedule 1 approved methods. Breeding of genetically modified mice was covered by project and personal licenses held by the last author. This study conformed to the ARRIVE guidelines.
For further methods, see details in the Appendix Methods.
Results
Evc mutants Have Frenulum and Vestibule Defects
To understand when Evc acts in dental and vestibula lamina development, LacZ expression in Evc heterozygous mice was followed using whole-mount X-Gal staining from E12.5, when both laminae start invaginating into the mesenchyme (Fig1A–C). In mice, the lower VL is prominent anteriorly and reduces posteriorly near the molars (Qiu and Tucker 2023); accordingly, LacZ was mainly expressed in the lower anterior vestibule and around the DL (Fig 1A–C). By E15.5, LacZ stain was prominent in the lower oral vestibule and mandible arch (Fig 1D), especially in the VL epithelium at a distance from the oral surface, and in the mesenchyme surrounding the forming tooth and VL (Fig 1E). The expression of this primary cilia gene is, therefore, highly specific during development.
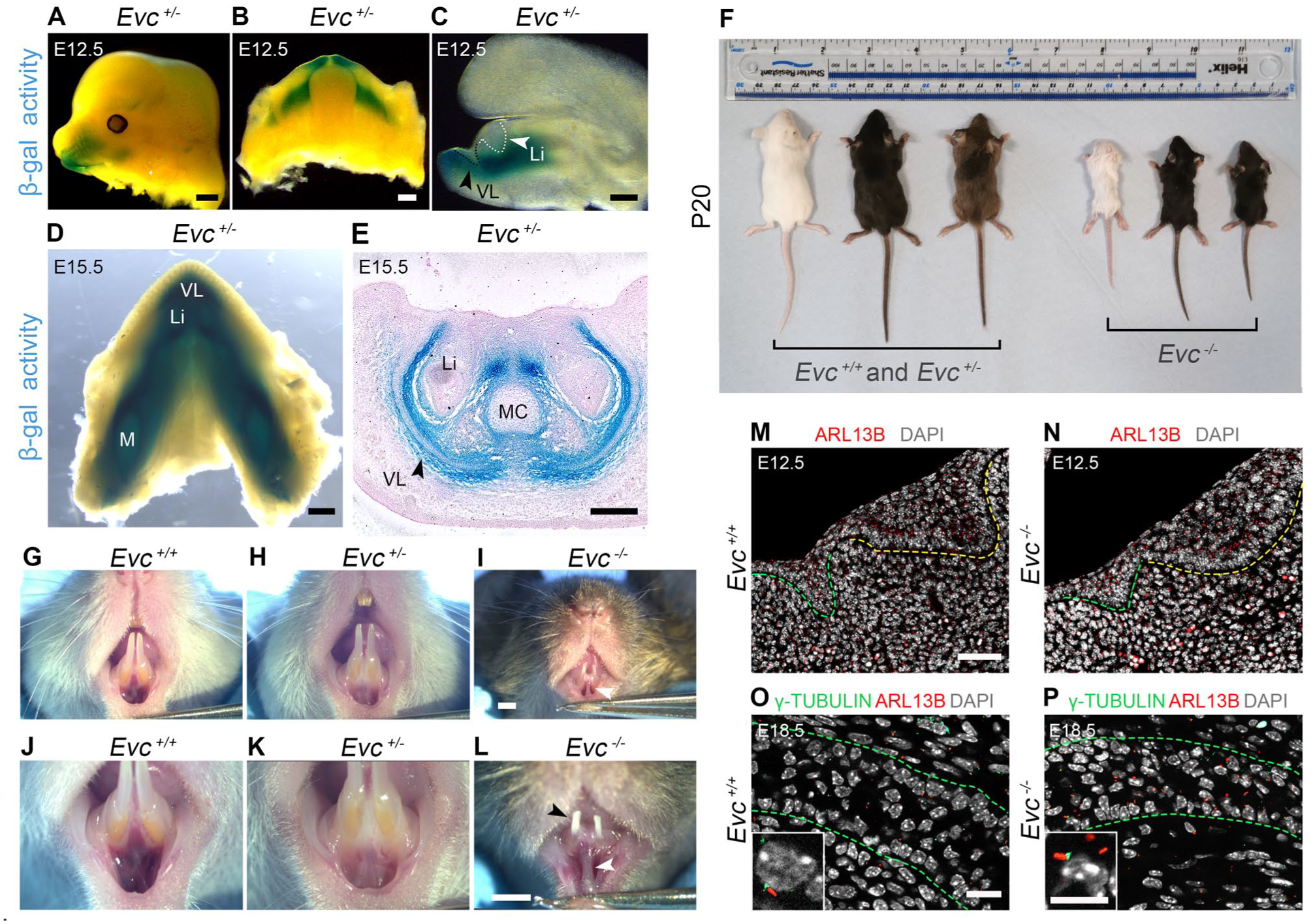
Loss of Evc leads to tooth and vestibule defects. (
Evc homozygous mutants (Evc mutants) survived to weaning but were smaller than their WT littermates at P20 (Fig 1F). At P20, Evc heterozygous mice did not show any marked physical differences (Fig 1F, H, K). Evc mutants had evident dental anomalies with hypoplastic incisors (black arrowhead, Fig 1I, L), consistent with previous findings (Nakatomi et al 2013). Notable defects were also seen in the oral vestibule, manifesting as a hyperplastic medial vestibule with high attachment to the gingival margin and midline frenulum (white arrowheads, Fig 1I, L).
Agreeing with data from the limb (Ruiz-Perez et al 2007; Caparros-Martin et al 2013), primary cilia were present in the mutant oral mesenchyme and epithelium at E12.5 and E18.5, as shown by expression of ARL13B and γ-tubulin, markers of the primary cilium membrane/axoneme and centrosome/basal body, respectively (Fig 1M–P).
Truncation of the VL in Evc Mutants Is Caused by Decreased Proliferation
Given the frenula and oral vestibule defects in juvenile mice, VL development was assessed in the anterior/incisor region, where the VL is most prominent in mice. At E13.5, 3-dimensional reconstructions revealed disrupted VL invagination around the incisor buds, with the VL nearly absent in the most anterior lower jaw (white arrowheads, Fig 2A), where the ectopic frenulum was observed postnatally (Fig 1L). By E14.5, Evc mutants displayed a significantly shallower VL when compared with the well-developed VL in the lower jaw of WT littermates (Fig 2B–E; for measurements see Appendix Fig 1; Appendix Table 1). To explore if the truncated VL was caused by reduced proliferation, we analyzed the incorporation of BrdU at E12.5 and E14.5. By E12.5, the VL and DL appeared as distinct E-cadherin–labeled thickenings, with significantly reduced proliferation in the mutant VL epithelium and surrounding mesenchyme (Fig 2F–I; Appendix Fig. 2; Appendix Tables 2 and 3). By E14.5, proliferation in the WT was concentrated on the buccal side and VL tip (Fig 2J, K) (Qiu and Tucker 2023), but this pattern was lost in mutants with reduced proliferation at the tip of the invaginating epithelium (Fig 2L–N; Appendix Fig 3; Appendix Table 4).
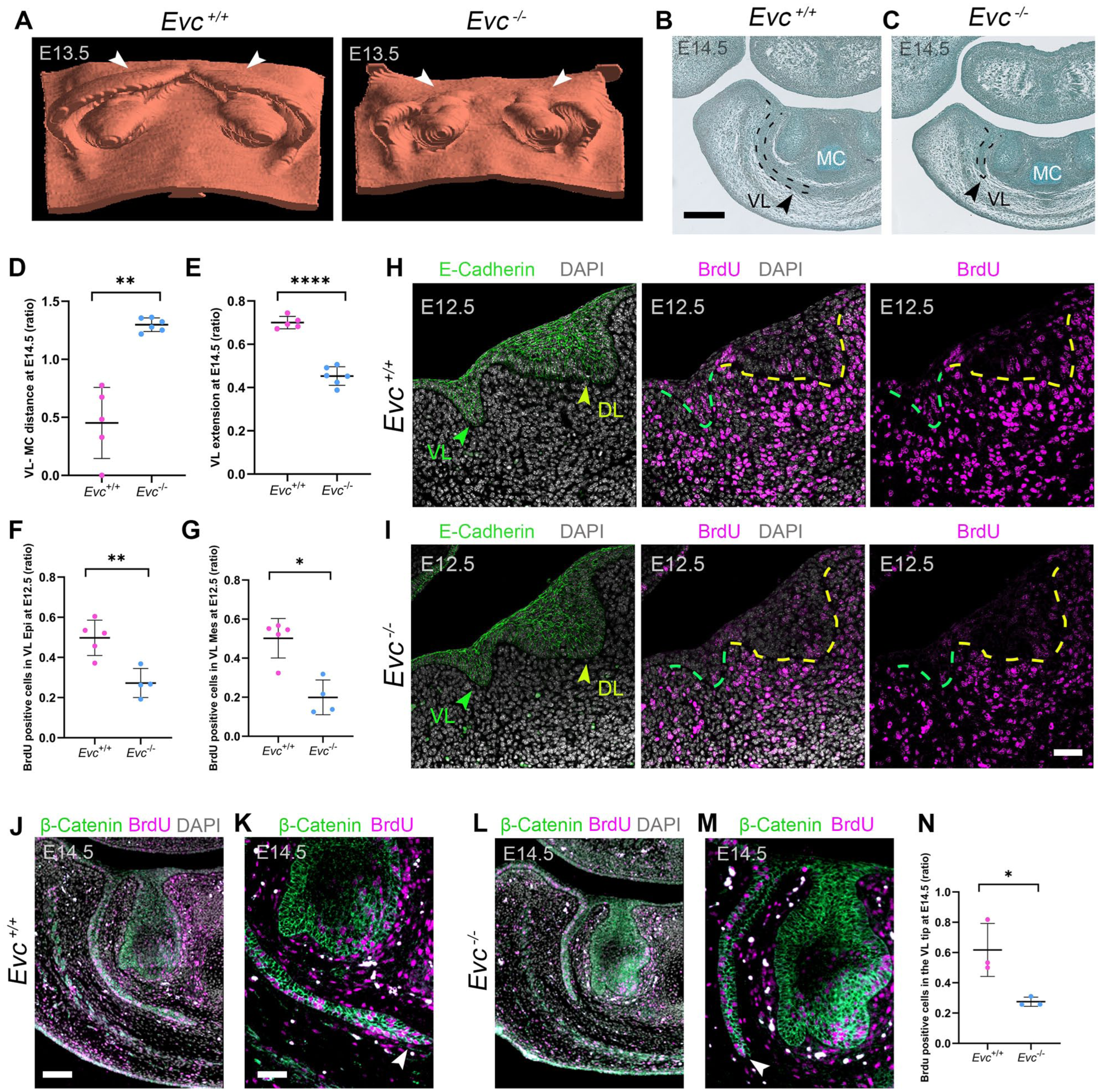
The VL is truncated in Evc mutants and shows impaired proliferation. (
Context-Dependent Changes in Gli1 Expression in Evc Mutants
Mutations in Evc or Evc2 impair Hedgehog signaling, as shown by reduced Gli1 or Ptch1 expression in the developing long bones of Evc and Evc2 mutants (Caparros-Martin et al 2013; Zhang et al 2015). A reduction in Hh signaling was also observed in the forming molars of Evc mutants (Nakatomi et al 2013) and developing incisors of Evc2 mutants (Zhang et al 2017). Gli1 reduction was also evident in Gas1 mutants, which have a truncated VL (Qiu et al 2023). We hypothesized Evc mutants would also show reduced Gli1 in the forming VL. At E12.5, as the VL starts to form, Gli1 expression is broadly expressed at the site of the dental placode (Hardcastle et al 1998) (Fig 3A). In Evc mutants, Gli1 was reduced in the tissue surrounding the VL and DL, with a significant downregulation in the mesenchyme around the VL (Fig 3A, B, E, F; Appendix Fig 4; Appendix Table 6). Downregulation of Shh signaling was also indicated by loss of Sostdc1, a target of Hh signaling in the dental mesenchyme (Cho et al 2011), which was highly expressed on the labial side of the forming VL in the WT and significantly downregulated in the mutant mesenchyme surrounding the VL (Fig 3C, D, G, H; Appendix Fig 4; Appendix Table 5).
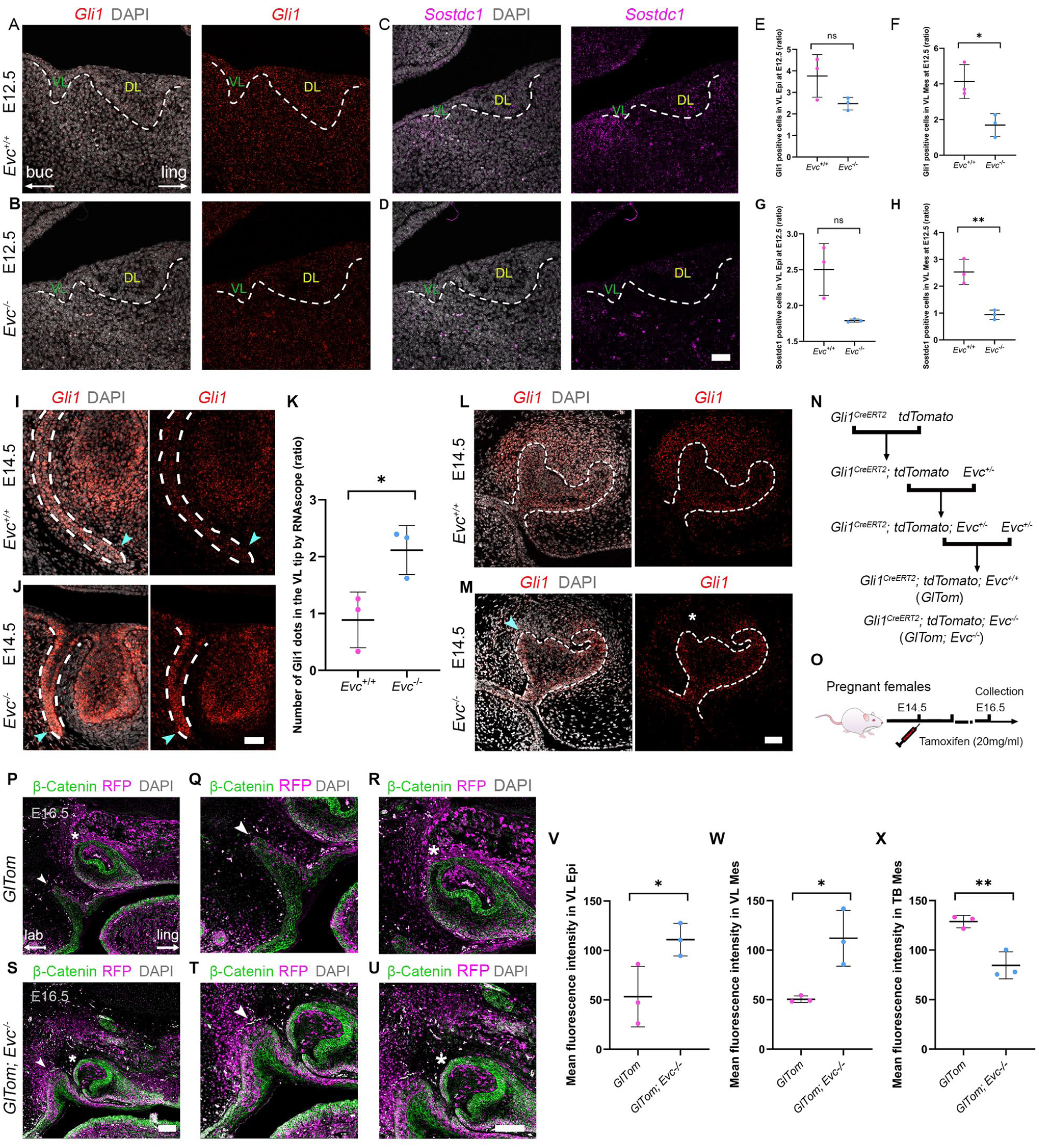
Tissue and time specific dysregulation of Shh signaling in Evc mutants. (
Interestingly, a different picture emerged from E14.5. At this stage, Gli1 was expressed in the VL epithelium but was absent from the very tip in WT mice (Fig 3I). In contrast, Evc mutants displayed high levels of Gli1 in the shortened VL, with expression concentrated at the tip (Fig 3J, K; Appendix Fig 5; Appendix Table 7). Ptch1, another readout of Hh signaling, was also upregulated in the mutant VL at E15.5, with no change in the expression of Shh (Appendix Fig 6A–D). By E18.5, Gli1 expression had turned off in the WT VL but remained high in mutants (Appendix Fig 6E, F). The impact of loss of Evc on Hedgehog signaling in the VL, therefore, appeared stage specific.
Given the surprising upregulation of Hedgehog targets in the VL at later embryonic stages, Gli1 expression was compared in the molar and incisor germs at this time point. As previously reported, Evc mutants exhibited buccal defects in the first molar at E14.5 (cyan arrowhead, Fig 3M) (Nakatomi et al., 2013), with reduced Gli1 expression in the surrounding mesenchyme (asterisk, Fig 3L, M). Consistent with a loss of Sonic Hedgehog signaling at the midline, upper incisors were frequently fused in mutants (Appendix Fig 7A–E, G–K), as seen in other Hedgehog pathway mutants (Seppala et al 2007). To corroborate these findings, we used Gli1CreERT2; tdTomato; Evc homozygous mice as a readout of Gli1 expression and lineage (Fig 3N). Tamoxifen was injected at E14.5, 2 d before sample collection (Fig 3O). Given the fusions, we focused on the upper incisor and neighboring VL. Interestingly, Gli1 lineage and expression were significantly reduced in the mesenchyme around the forming incisor but were upregulated in the mesenchyme and epithelium of the adjacent VL (Fig 3P–X; Appendix Fig 8; Appendix Table 8, 9). The developing dentition and VL, therefore, displayed opposing Hedgehog signaling responses from E14.5 onward.
The Short VL Opens as Normal to Create a Shallow Furrow
The murine VL differentiates and opens postnatally in the mouse to create the vestibule (Qiu et al 2023). Ectopic frenula could be created by either failed VL invagination or defective VL opening. We therefore analyzed mutant VL postnatally to assess the lamina opening. At early postnatal stages, the VL in WT mice had undergone further extension, leading to the convergence of the two laminae at the midline (Fig 4A–D). In contrast, Evc mutants consistently demonstrated a shortened VL (Fig 4E–H). Interestingly, despite the shallow invagination, the Evc mutants displayed a comparable VL opening pattern to WT littermates, with the VL initiating opening by P5 (Fig 4B, F) and achieving full patency by P13 (Fig 4D, H) (n = 6/6, P5–P13). Differentiation progressed as normal, as indicated by expression of E-cadherin, occludin, and loricrin (Fig 4I–R). Interestingly, postnatally in a subset of mutants, the VL was observed to undergo additional outpocketings (black arrowheads) that opened to form side canals, not present in WT (n = 7/12 mutants, Fig 4G, H), suggesting not only truncation but also abnormal branching of the VL.
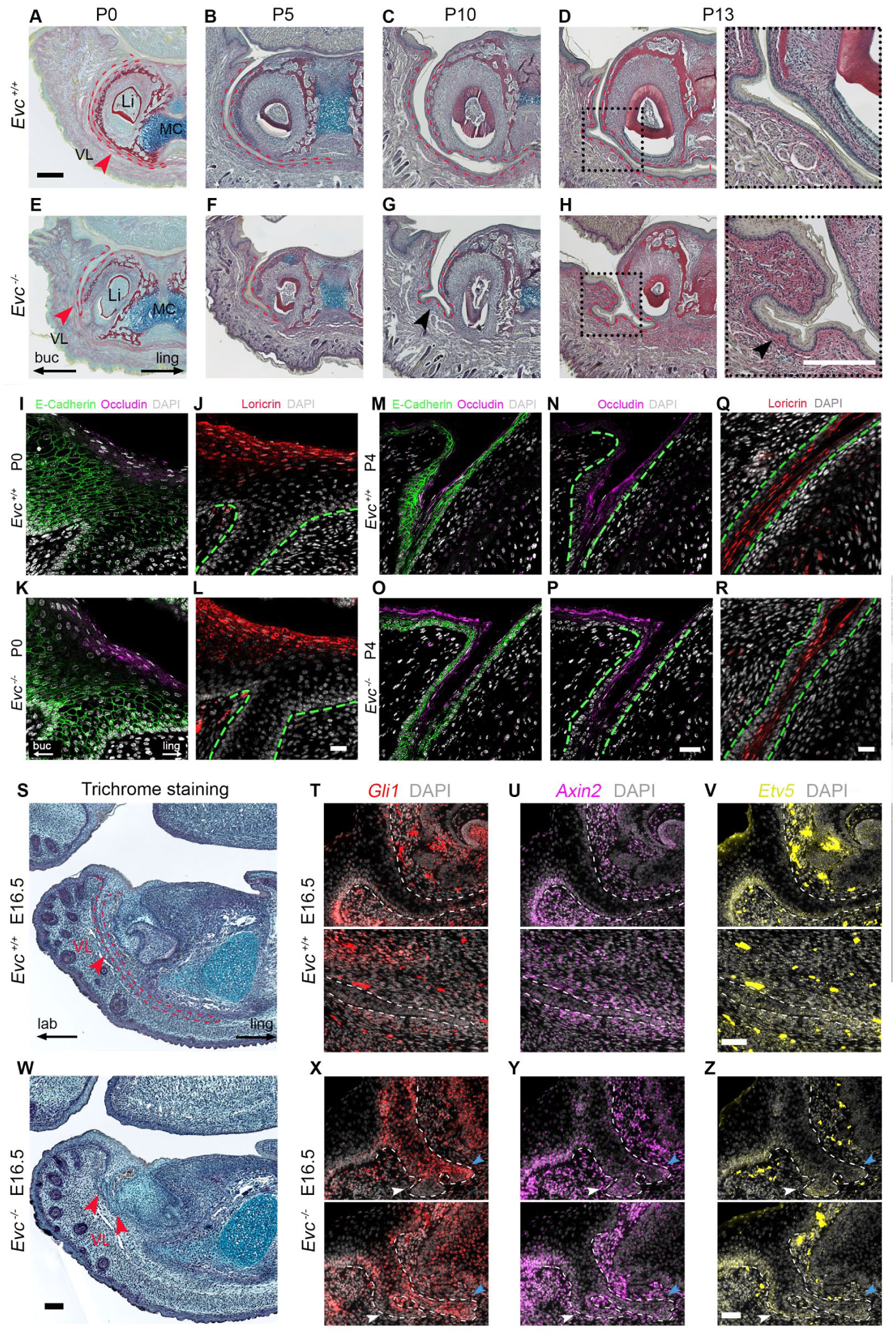
Evc mutants presented shortened VL along with the normal opening of the VL and changes in VL fate linked to branching. (
Changes in VL Fate Linked to Branching and Ectopic Tooth Formation
In addition to VL defects and incisor fusions, Evc mutants exhibited a range of dental abnormalities, including malformed teeth, hypodontia, and fused upper molars (Fig 1L; Appendix Fig 7F, L). Of particular interest was the formation of supernumerary teeth (n = 4/26 mutants). These extra teeth were located near the upper incisor at P10 and P20 (Appendix Fig 9), and in the lower incisor region (Fig 5B). The VL has latent odontogenic potential that can be reawakened under certain conditions (Hovorakova et al 2020). We therefore examined whether the VL shifts fate to form ectopic tooth germs.
At E16.5, the WT VL had elongated beneath Meckel’s cartilage, encircling the developing incisor germ as a thin sheet of epithelium (Fig 4S), while in several samples, the shortened VL of Evc mutants had branched, thereby creating 2 to 3 additional epithelial structures (n = 2/4 mutants, Fig 4W). Overall, branched vestibular laminae were observed in 5 of 14 mutant prenatal samples, which may explain the origin of the side branches of the VL observed postnatally.
To better understand branched vestibular laminae, we compared readouts of Hedgehog signaling with that of Wnt and Fgf, which interact during morphogenesis in many developing organs. In WT vestibular laminae, the invaginating epithelial tip of the lamina had a distinct molecular profile compared with the rest of the lamina. The tip was characterized by low Gli1, high Axin2, a readout of canonical Wnt signaling (Lohi et al 2010), and high Etv5, a member of the family of signal-regulated transcription factors induced by Fgf signaling (Garg et al 2020) (Fig 4T–V; Appendix Fig 10A–C). Interestingly, the different branches of the VL in the Evc mutants displayed distinct molecular profiles. The labial (lip side) VL (white arrowheads) mirrored the WT VL at the tip, while the lingual (tongue side) branches (cyan arrowheads) exhibited co-expression of Gli1 with Etv5 and Axin2 (Fig 4X–Z; Appendix Fig 10D–F). This suggested a change in fate of the cells of the VL on the lingual side.
Interestingly, in one specimen at E18.5, we identified the formation of an ectopic tooth germ near the lingual branch of the VL in the mutant (Fig 5A, B). The effect was unilateral, with the contralateral VL being truncated but not associated with a branched VL (Fig 5B). Stimulation of Wnt signaling in the oral epithelium has been shown to be key for stimulating tooth development (Popa et al 2019). We therefore investigated whether Wnt signaling was upregulated in the VL forming the additional tooth by examining nuclear β-catenin. Nuclear localization and increased expression of β-catenin was observed in mutant VL when compared with the WT (Fig 5C, D), suggesting increased activity of the canonical Wnt signaling in the mutant. Elevated Wnt expression can suppress SOX2 in a tissue-dependent manner (Popa et al 2019). Consistent with this, SOX2 expression was reduced in the tip of the mutant VL compared with WT (Fig 5E, E′). Moreover, expression of LEF1, a target of Wnt signaling, was elevated in the epithelium of the mutant VL at E15.5 (Fig. 5F, F′). Thus, mutations in Evc can lead to increased activity of the Wnt signaling pathway in the aberrantly branched VL epithelium.
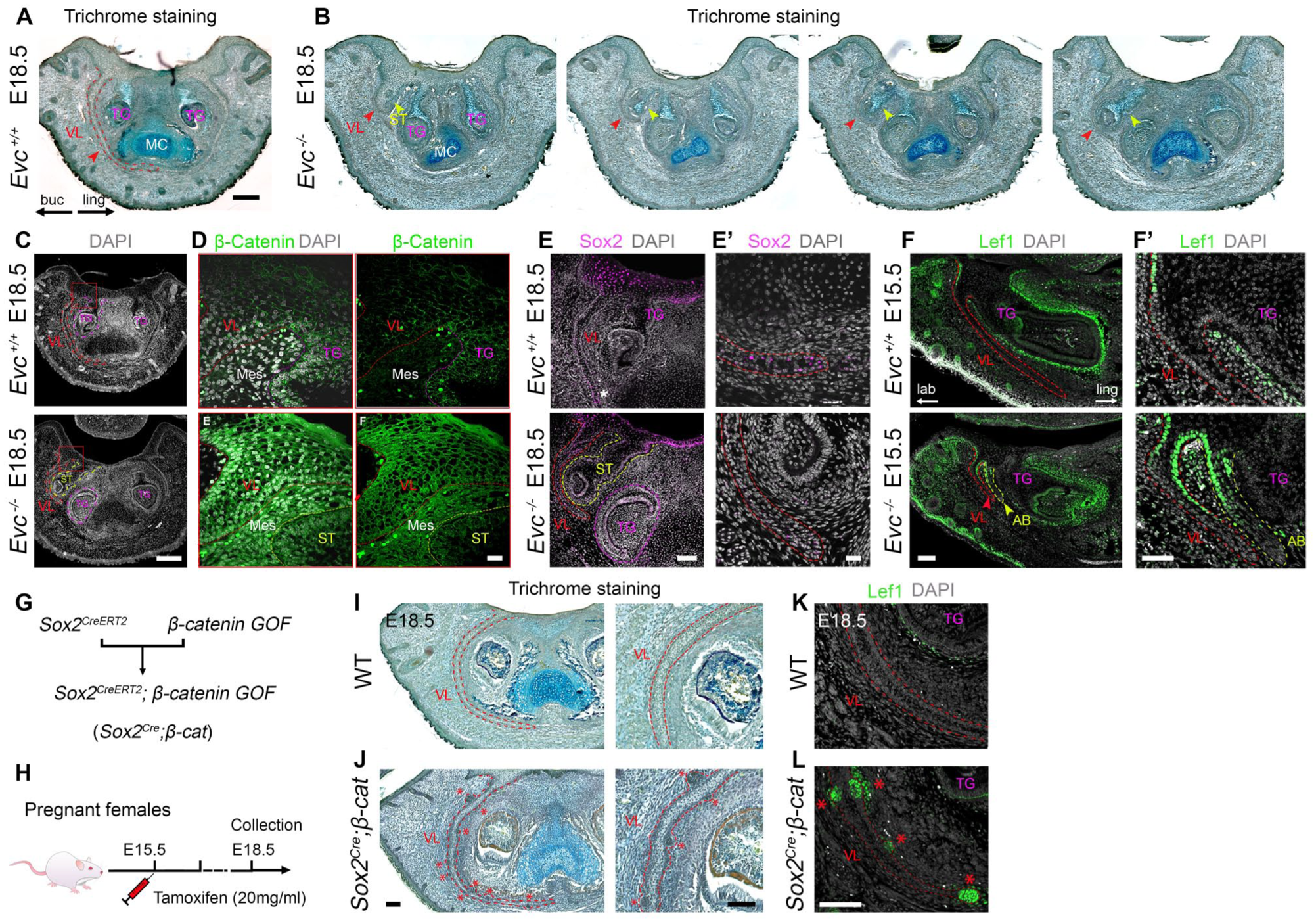
Enhanced Wnt signaling and ectopic tooth germs in Evc mutants. (
Enhanced Wnt/β-Catenin Signaling in the VL Recapitulates the Formation of Ectopic Tooth Buds
To investigate if upregulated Wnt signaling in Evc-mutant lamina was sufficient to drive supernumerary tooth formation, we activated canonical Wnt/β-catenin signaling in the VL using Sox2 CreERT2 ; β-catenin GOF mice (Fig 5G). SOX2 is strongly expressed in the VL from early stages of development (Appendix Fig 11). Tamoxifen was administered to pregnant β-catenin GOF mothers mated to Sox2CreERT2 males at E15.5, and embryos were collected at E18.5 (Fig 5H). In keeping with the formation of ectopic teeth near the lamina in Evc mutants, small, toothlike structures were observed protruding from the VL in the Sox2CreERT2; β-catenin GOF mice (red asterisks, Fig 5I, J). These toothlike structures displayed mesenchymal condensation and expressed LEF1, thus mirroring the characteristic “early bud stage” observed in typical tooth formation (n = 3/3, red asterisks, Fig 5K, L). The upregulated canonical Wnt signaling in the Evc mutant VL is therefore likely to be a key step in the formation of ectopic tooth germs in this region.
Discussion
Oral Vestibule Defects in Evc Mutants Are Driven by an Early Loss of Proliferation and Not Defects in Opening of the Lamina
Vestibular defects are common in EvC syndrome patients, marked by multiple frenula and gum attachments to the lips and cheeks (Sasalawad et al 2013). These could result from impaired VL invagination or defects in VL opening. Previously, the long bones in Evc mutants have been shown to display normal proliferation but a change in the timing of differentiation with premature formation of hypertrophic chondrocytes (Ruiz-Perez et al 2007), while molars exhibited reduced proliferation, especially on the buccal/cheek side, during cap and bell stages (Nakatomi et al 2013). We show that Evc mutant embryos have consistently shorter vestibular laminae when compared with WT littermates but with normal differentiation leading to postnatal opening (N = 26/26). Our analysis highlights an early defect in proliferation in the mesenchyme and epithelium as the driver. The shallow vestibule defects in patients are therefore likely to be due to defects in early invagination due to proliferation changes rather than opening defects due to abnormal differentiation.
Tissue- and Time-Specific Impact of Loss of Evc
Defects in Evc mutants, including those in long bones and molars, are associated with downregulation of Hedgehog signaling with reduction of Ptch1 and Gli1 (Ruiz-Perez et al 2007; Nakatomi et al 2013). Consistent with loss of Hedgehog signaling, we observed midline defects with fused upper incisors and molars (Appendix Fig 7) (Seppala et al 2007; Asrar et al 2025). Loss of Gas1 has previously been shown to lead to reduced Gli1 and a truncated VL (Qiu et al 2023), and similarly, Gli1 was significantly downregulated in the mesenchyme of the Evc mutant VL at initiation. Short vestibular laminae are therefore associated with low Hedgehog signaling, particularly in the mesenchyme correlating to regions of high Evc expression. This agrees with findings that the addition of cyclopamine leads to shorter vestibular laminae in explant culture (Qiu et al 2023).
From E14.5 onward, some Evc vestibular laminae underwent aberrant branching (N = 12/26) with infrequent formation of supernumerary teeth (N = 4/26). At these stages, the mutant VL was associated with increased and persistent Gli1 and Ptch1 expression compared with WTs, suggesting distinct pathological mechanisms driving the short versus branching vestibular laminae.
Although primary cilia are structurally conserved and functionally uniform, defects in the same ciliary machinery can lead to distinct effects on different tissues (Elliott and Brugmann 2019; Brooks et al 2021). This may reflect tissue-specific balances of GLIR (GLI repressor) and GLIA (GLI activator) forms, with varying GLIR thresholds needed to prevent derepression-associated phenotypes (Hwang et al 2023). These differences in GLI forms may therefore explain the differences in response of the early and late developing VL and neighboring tooth germs to the same mutation.
Branching and Fate Changes of the VL in Evc Mutants
All Evc mutants had short vestibular laminae, but only some branched or displayed ectopic teeth. Similarly, EvC syndrome patients may not show the full spectrum of symptoms even among patients with the same mutation (Ulucan et al 2008). The variability shown in our mouse mutants may be due to their mixed genetic background (CD1/C57BL/6), as pure C57BL/6 mutants did not survive postnatally. Ectopic teeth not associated with the VL were also evident near the upper incisor, suggesting two independent mechanisms for supernumerary tooth formation in patients.
The labial and lingual branches of the mutant VL showed different patterns of expression for readouts of Hh, Fgf, and Wnt signaling. Fgf signaling is key to branching in many epithelial tissues (Jones et al 2020), and abnormal expression in the lamina may cause the lamina to divide. The Fgf pathway has also been linked to cilia defects, with Sprouty mutants displaying features of ciliopathies (Hruba et al 2021).
Epithelial Wnt overactivation can trigger additional tooth germs, potentially leading to odontomas (Popa et al 2019). Hh and Wnt signaling pathways are known to interact closely in various tissues, and alterations in Hh signaling can affect Wnt signaling, through misregulation of Sostdc1 (Sarkar et al 2000; Cho et al 2011; Kumar et al 2021). Elevated expression of Wnt signaling was previously reported in Evc2 mutant molars (Zhang et al 2022). Here we show early downregulation of Sostdc1 and an upregulation of canonical Wnt signaling in mutant VL epithelium, potentially explaining the formation of supernumerary tooth germs. Supporting this, Wnt activation in Sox2+ VL cells was shown to induce ectopic tooth buds, confirming VL’s tooth-forming potential under pathological conditions. The downregulation of Sostdc1 in the mutant presents an interesting avenue for further investigation as it may be possible to rescue some of the VL defects by addition of Sostdc1 to reset the balance of Wnt signaling in the absence of normal Hedgehog signaling.
In conclusion, the investigation of VL and dental defects in Evc mutants offers novel insights into the underlying mechanisms involved in ciliopathies, particularly EvC syndrome. These findings highlighted that closely developing structures can respond very differently to the same mutation with both loss and overexpression of Gli1. Similar vestibular frenula defects, particularly a short frenulum (frenulum breve), are associated with other ciliopathies such as oral-facial-digital syndrome (Bruel et al 2018) and Joubert syndrome (Penon-Portmann et al 2022), suggesting that shared pathological mechanisms may underlie across various ciliopathies.
Author Contributions
T. Qiu, contributed to conceptualization, methodology, validation, formal analysis, investigation, data curation, visualization, funding acquisition, drafted the manuscript, and critically revised the manuscript; M. Hovorakova, H. Peters, contributed to resources, methodology, and critically revised the manuscript; A.S. Tucker, contributed to conceptualization, resources, methodology, data curation, supervision, funding acquisition, project administration, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345261430674 – Supplemental material for Aberrant Proliferation and Cell Fate Underlie Oral Defects in a Mouse Model of EvC Syndrome
Supplemental material, sj-docx-1-jdr-10.1177_00220345261430674 for Aberrant Proliferation and Cell Fate Underlie Oral Defects in a Mouse Model of EvC Syndrome by T. Qiu, M. Hovorakova, H. Peters and A.S. Tucker in Journal of Dental Research
Footnotes
Acknowledgements
We thank Victor Luis Ruiz-Perez (CSIC-UAM - Instituto de Investigaciones Biomédicas Biomedicas Sols-Morreale (IIBM)) for providing the Evc mouse line and Herve Lesot (IAPG, Brno, Czech Republic) for assistance with 3-dimensional reconstructions of the VL.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by grants from the BBSRC (BB/W00240X/1 to A.S. Tucker) and Grant Agency of the Czech Republic (21-04178S to A.S. Tucker), with additional support from Charles University (Cooperation project 207036-10 Morphological disciplines of medicine/LF1 MH), and the China Scholarship Council as part of a PhD studentship at KCL (T. Qiu).
Data and Materials Availability
A supplemental appendix to this article is available online.