
Abstract
Diabetes mellitus (DM) and periodontitis share a complex, bidirectional relationship, with each condition exacerbating the other. Diabetes, particularly when poorly controlled, significantly increases the risk, severity, and progression of periodontitis. The biological mechanisms involved are complex and numerous. Hyperglycemia in diabetes is linked to oral microbial dysbiosis, which is in turn associated with increased inflammation, epithelial barrier dysfunction, impaired neutrophil and macrophage function, altered T-cell profiles, and cytokine imbalance, collectively fostering chronic inflammation and immune dysregulation. Moreover, diabetes alters bone metabolism, promoting osteoclastogenesis and reducing reparative bone regeneration by limiting coupled bone formation through an effect on growth factor production, mesenchymal stems cells, and osteoblasts. Conversely, periodontitis is strongly linked to poor glycemic control. Clinical studies and longitudinal meta-analyses report consistent positive associations, while randomized controlled trials show that periodontal therapy reduces HbA1c by ~0.43%. Emerging evidence suggests that periodontitis and oral preclinical dysbiosis contribute to diabetogenesis, although causality remains uncertain. Periodontitis may drive metabolic dysfunction through several biological mechanisms. The dysbiotic oral microbiome and subsequent periodontitis may promote systemic inflammation and subsequent insulin resistance and glucose intolerance. Moreover, oral dysbiosis may deplete nitrate-reducing taxa and impair nitric oxide pathways, which has relevance to both periodontal and cardiometabolic health. Accordingly, periodontal treatment in diabetic populations has shown potential health care savings. Nevertheless, trials assessing the influence of periodontitis treatment on systemic outcomes consistently show significant treatment heterogeneity, which requires explication in future studies. This review underscores the systemic implications of periodontitis in diabetes and highlights the value of integrating periodontal care into diabetes management. A better understanding of the shared pathophysiology between these diseases supports interdisciplinary approaches and points toward novel preventive and therapeutic strategies targeting inflammation, microbial balance, and host response modulation to jointly benefit periodontal and cardiometabolic health.
Introduction
Diabetes mellitus (DM) and periodontitis are associated. Substantial evidence suggests that these associations are bidirectional and causal (Lalla and Papapanou 2011; Nascimento et al 2018; Graves et al 2020; Stohr et al 2021). Most patients with diabetes fall into two broad pathogenetic categories, type 1 DM (T1DM) and type 2 DM (T2DM). T1DM is characterized by a lack of insulin due to a destruction of pancreatic beta cells. In contrast, decreased insulin action is the basis for T2DM, the most common form of DM, accounting for 90% to 95% of DM worldwide. In both forms of DM, poor management manifests as chronic hyperglycemia, which leads to complications including cardiovascular disease, kidney disease, neuropathy, blindness, and impaired wound healing that can lead to lower-extremity amputation. DM also affects oral health, with increased risk and severity of periodontitis (Lalla and Papapanou 2011; Graves et al 2020).
Early research often demonstrates that diabetes is associated with increased periodontitis, although evidence mainly derives from observational and animal studies. Conversely, periodontitis worsens glycemic control among diabetic individuals, with periodontal treatment showing beneficial effects. Emerging evidence further links oral dysbiosis from periodontitis to T2DM development, suggesting a cyclical relationship. Given the projected rise of diabetes prevalence from 37.3 million in 2019 to 60.6 million by 2060 in the United States alone (Lin et al 2018; Stohr et al 2021), understanding this relationship is crucial. This review explores these mechanisms (Fig 1).
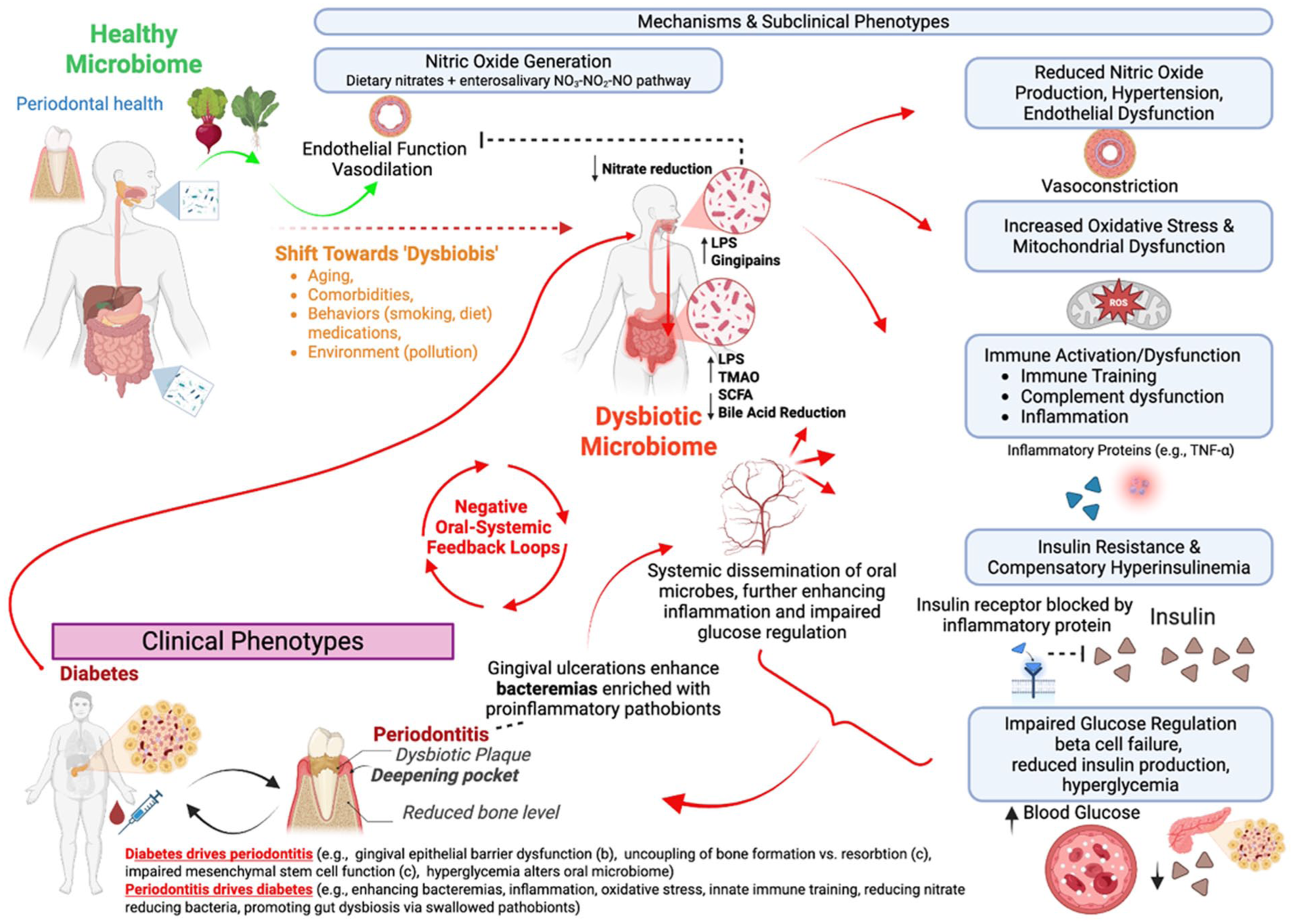
Bidirectional relationship between periodontitis and diabetes mellitus. Periodontal disease is associated with active immune and inflammatory responses that are related to the changes in the oral microbiome. These responses participate in a feedback loop between periodontitis and hyperglycemia due to diabetes mellitus, related to either insulin deficiency or insulin resistance. In addition, adipose tissue participates in this bidirectional inflammatory process through the secretion of proinflammatory cytokines, higher oxidative stress, and disruption of signaling pathways.
Diabetes as a Contributor to Periodontitis
Diabetes is recognized as a causal risk factor for periodontitis, supported by observational and animal studies (Lalla and Papapanou 2011; Graves et al 2020). Longitudinal studies, such as the Study of Health in Pomerania (SHIP) study, have reported that uncontrolled diabetes exacerbates periodontal attachment loss more significantly than controlled diabetes does (Demmer et al 2010). However, clinical trials remain scarce, limiting precise causal estimation. Current observational research often lacks rigorous confounder adjustments and standardized definitions of periodontitis, creating validity challenges. Despite these limitations, diabetes clearly affects periodontal health, warranting further investigation to identify effective interventions.
Periodontitis and Risk for Incident T2DM
Historically, diabetes-induced pathophysiology was considered the primary driver linking diabetes and periodontitis. However, recent evidence indicates oral dysbiosis in periodontitis may independently contribute to incident T2DM, broadening the bidirectional hypothesis. A meta-analysis of 10 studies reported a significant association between periodontitis and incident diabetes (risk ratio = 1.26 [95% CI, 1.12 to 1.41]) (Stohr et al 2021). Yet, causality remains uncertain due to a lack of large-scale intervention trials, as classical randomized interventions would be extremely resource intensive and likely unethical due to the requirement of untreated control groups. Future trials targeting prediabetic populations would be informative and more feasible to clarify this relationship.
Periodontitis, Insulin Resistance, and Prediabetes among Diabetes-Free Individuals
At least 6 studies have reported associations between periodontitis and prediabetes, beginning with Saito et al (2004), who linked periodontal status to incident impaired glucose tolerance. The American Diabetes Association describes prediabetes as blood glucose levels that are higher than normal but not high enough to meet the criteria for diabetes (Demmer 2015). Subsequent studies confirmed associations with impaired fasting glucose, impaired glucose tolerance (Arora et al 2014; Adam et al 2024), and elevated HbA1c. Longitudinal data also link clinical attachment loss to rising HbA1c levels (Demmer et al 2010) and insulin resistance. The ORIGINS study expanded this by associating subgingival microbiome composition with inflammation, insulin resistance (Demmer et al 2017), prevalent prediabetes (Demmer et al 2015), and fasting glucose changes (Demmer et al 2019) in diabetes-free individuals. A single-arm periodontal treatment study among patients with prediabetes reported notable A1c reductions following treatment but lacked a randomized comparator group (Kocher et al 2019). On balance, these relationships are supported by meta-analyses of the relationship between periodontitis and metabolic syndrome more broadly (Pirih et al 2021). Nevertheless, as noted above, existing studies suffer from residual confounding, possible selection bias, and/or lack of temporality, and there are no clinical trials testing whether periodontal interventions prevent insulin resistance or prediabetes development.
Periodontal Treatment and Glycemic Control in Patients with Diabetes
Robust evidence supports periodontal therapy for glycemic control improvement in diabetic patients. A recent systematic review reported a clinically meaningful 0.43% reduction in HbA1c within 3 to 4 mo posttreatment among individuals receiving diabetes medications (Simpson et al 2022), comparable to monotherapy effects (Feingold 2000). Heterogeneity (I² = 60% to 80%) remains substantial (Teeuw et al 2010; Oliveira et al 2023), necessitating further research. Network meta-analyses highlight adjunctive antimicrobials such as doxycycline gel (−0.80%), chlorhexidine gel (−0.68%), and tetracycline fiber (−0.62%) as enhancing HbA1c reductions (Marchiori Sant’Anna Leal de Oliveira et al 2024) (Fig 2). In addition, periodontal interventions potentially yield substantial health care savings (Saenz-Ravello et al 2024). Future research focusing on mechanisms underlying heterogeneity may lead to targeted therapies improving clinical outcomes and cost-effectiveness.
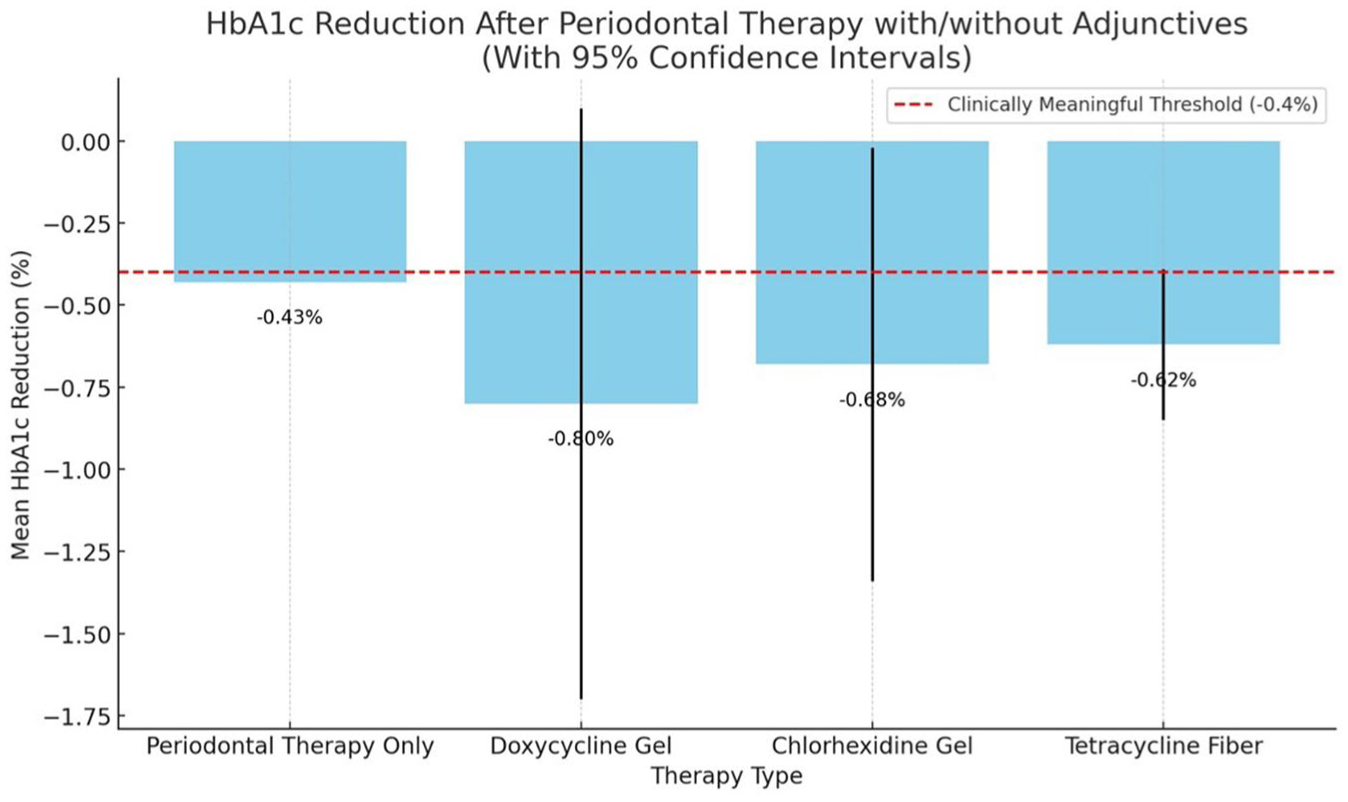
Periodontal treatment and glycemic control in patients with diabetes. The graph demonstrates the average HbA1c reduction from periodontal therapy and adjunctive antimicrobial treatments, along with their 95% confidence intervals. The red dashed line indicates the threshold for a clinically meaningful change (−0.4%).
Mechanisms Connecting DM to Periodontitis Development and Progression
Oral Microbiome Alterations
DM induces significant changes in the oral microbiota. In T1DM, microbial communities show an increased abundance of Streptococcus, Veillonella, and Lactobacillus genera, alongside upregulation of genes involved in transport and metabolic pathways (Longo et al 2018; Graves et al 2019; Carelli et al 2023; Ebersole et al 2025). In contrast, T2DM is characterized by a depletion of Fusobacteriota and Campilobacterota phyla and an enrichment of Proteobacteria, suggesting a shift toward proinflammatory and potentially pathogenic communities (Longo et al 2018; Graves et al 2019, 2020; Ebersole et al 2025). These compositional shifts vary between studies, and there is currently no consensus on the degree of dysbiosis induced by hyperglycemia. Despite variability across studies, hyperglycemia-induced inflammation appears to be a consistent driver of these microbial shifts. Elevated gingival crevicular fluid proteins and increased expression of inflammatory mediators such as interleukin (IL)–1β, matrix metalloproteinase (MMP)–8, and MMP-9 are observed in diabetic patients, correlating with the altered microbiota (Ebersole et al 2025). Diabetes-related inflammation is noteworthy as it alters the oral microbiota. The development of diabetes in susceptible mice coincides with significant shifts in oral microbial profiles (Xiao et al 2017). The inhibition of inflammation with IL-17–specific antibodies significantly attenuates the pathogenicity of the oral microbiota caused by diabetes (Xiao et al 2017), suggesting that diabetes-enhanced inflammation increases bacterial pathogenicity in vivo.
Epithelial Barrier Dysfunction
Diabetes compromises gingival epithelial barrier integrity through multiple pathophysiological mechanisms. Hyperglycemic conditions impair keratinocyte migration and activity through the upregulation of MMP-9 and other factors, with significant contributions from epigenetic modifications (Yang et al 2024). High glucose exposure induces senescence marker expression (p16, p21) in gingival keratinocytes while simultaneously reducing essential junctional proteins including Claudin-1, E-cadherin, and Connexin 43, indicative of a compromised barrier function (Zhang et al 2021). Diabetes may also inhibit autophagy and promote senescence, thereby compromising the structural integrity of gingival epithelial cells (Liu et al 2024; Ye et al 2024). Metformin reverses hyperglycemia-induced barrier dysfunction and may enhance epithelial structural integrity through reduced senescence (Ye et al 2024). In diabetic murine models, the loss of epithelial junction proteins (E-cadherin, Claudin-1) correlates with enhanced neutrophil extracellular trap (NET) formation. The inhibition of NET accumulation restores epithelial barrier function and reduces periodontal tissue destruction (Wang et al 2024) (Fig 3).
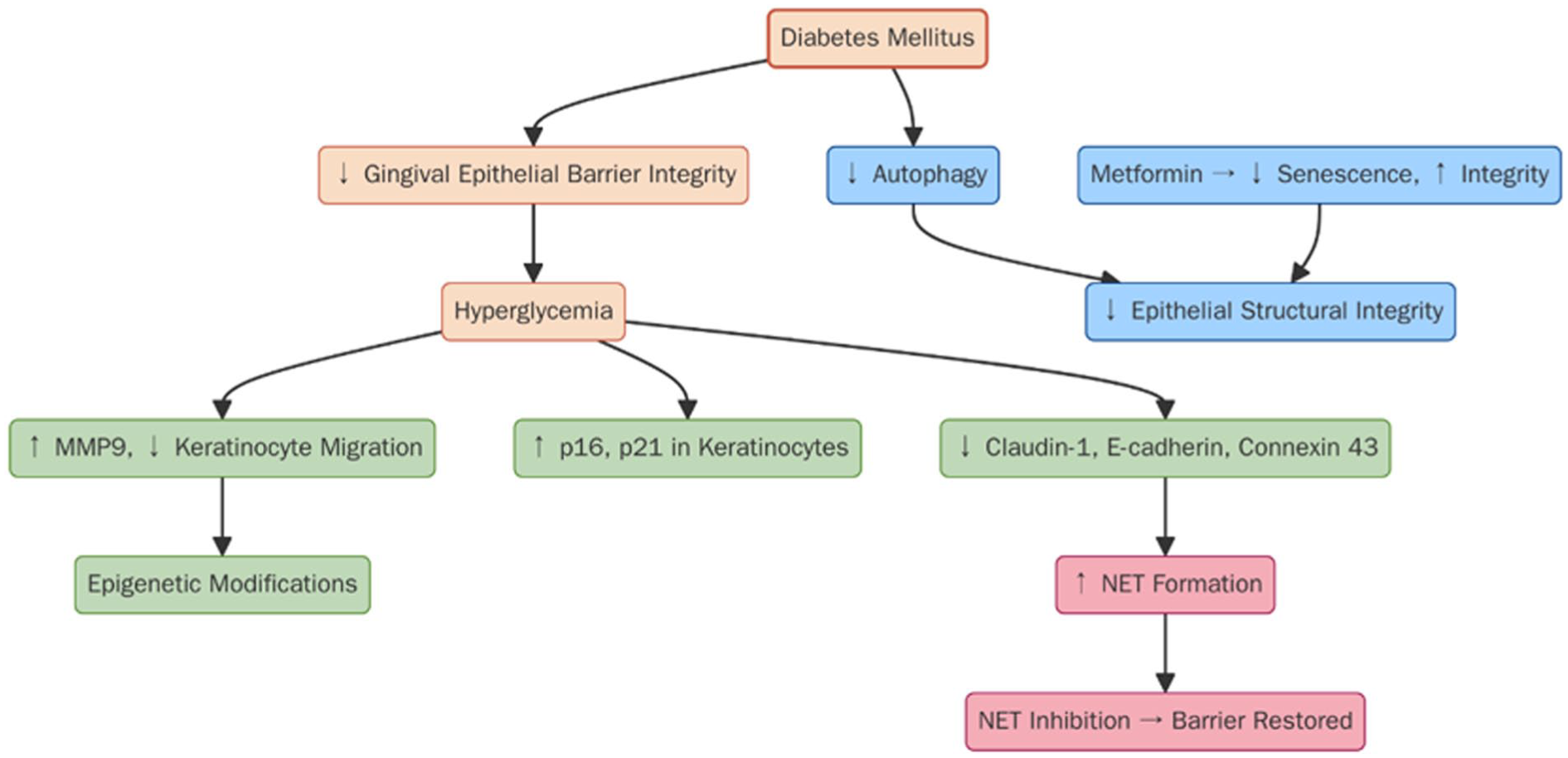
Diabetes-induced gingival epithelial barrier dysfunction. Diabetes mellitus impairs gingival epithelial integrity via multiple mechanisms, including hyperglycemia-induced upregulation of lytic enzymes such as matrix metalloproteinase–9 (MMP9), reduced keratinocyte function, epigenetic modifications, and increased senescence. Hyperglycemia and reduced autophagy and neutrophil extracellular trap (NET) formation may further lower junctional protein expression (eg, claudin-1, E-cadherin, connexin 43). Metformin treatment improves barrier function by reducing senescence and enhancing epithelial integrity, linking both to hyperglycemia or the direct effect of this drug.
Dysregulated Host Immune Response
Diabetes affects the periodontium by increasing and prolonging inflammation, which leads to increased tissue destruction that can be inhibited by blocking tumor necrosis factor (TNF) (Andriankaja et al 2012). Neutrophils, which play a key role in antibacterial defense, undergo hyperactivation and dysregulation in diabetic environments, resulting in greater tissue damage. Diabetes also renders neutrophils less effective, impairing bacterial clearance. A contributing factor is the impact of diabetes on decreased CXCL1 expression in gingival fibroblasts that stimulate neutrophil responses. Targeted CXCL1 overexpression in gingival tissues normalizes immune cell recruitment and prevents diabetes-enhanced alveolar bone loss (Shinjo et al 2023). Single-cell RNA sequencing and flow cytometry reveal that diabetes dysregulates lymphocytes disrupting the immune balance, notably increasing IL-17a+ γδ T cells and reducing regulatory T cells in humans and mice and promoting proinflammatory neutrophil polarization in the gingival tissue (Alghamdi et al 2025). Selectively inhibiting γδ T cells that express IL-17a reverses periodontal damage, highlighting these cells as key drivers of diabetes-enhanced periodontal tissue destruction.
Diabetes enhances proinflammatory cytokine (IL-1, TNF) release from macrophages, exacerbating periodontal tissue destruction by polarizing macrophages to an M1 phenotype with concurrent reduction in reparative M2 populations (Wu et al 2025). Recent investigations have elucidated the complex effects of T2DM on adaptive immunity, particularly in the context periodontitis ranging from early (stage 1) to severe (stage 4) and interactions with metabolic disorders such as obesity. T2DM elevates Th1 and Th17 cell populations, intensifying inflammatory responses and periodontitis severity, as evidenced by increased fasting plasma glucose, glycated hemoglobin, interferon (IFN)–γ, and IL-17 levels that correlate with deteriorating periodontal indices. Hyperglycemic conditions disrupt immunoregulatory mechanisms in vitro through cytokine profile alterations, notably upregulating IL-6 and IL-23 while downregulating IL-10. The interrelationship between diabetes, obesity, and periodontitis involves unique immunological mechanisms, where diabetes-primed B cells exacerbate periodontal degradation, particularly in obese or insulin-resistant individuals (Zhu et al 2014). Diabetic conditions enhance T-cell–mediated responses, resulting in increased alveolar bone loss following bacterial infections. Enhanced IFN-γ expression in diabetic mice similarly links Th1-driven responses to alveolar bone loss in periodontitis. In addition, T2DM influences dendritic cell function, promoting Th1 and Th17 lymphocyte generation while reducing regulatory T cell (Treg) populations, leading to unresolved inflammation (Guzman-Flores et al 2020; Alghamdi et al 2025). These findings collectively illustrate the complex interplay between hyperglycemia, immune dysregulation, and their implications for periodontal and systemic health in T2DM patients.
Bone Metabolism Disruption
Diabetes impairs bone homeostasis through multiple mechanisms, enhancing osteoclastic activity and reducing bone repair. Chronic hyperglycemia promotes osteoclastogenesis and impairs osteoblast function via increased proinflammatory cytokines and RANKL expression (Graves et al 2020). Insulin counteracts these effects by restoring the OPG/RANKL ratio. The role of periodontal ligament (PDL) fibroblasts, osteoblasts, and osteocytes in periodontitis-related bone loss is shown by reduced tissue destruction in mice with lineage-specific nuclear factor–κB (NF-κB) inhibition (Zheng et al 2018). Diabetes disrupts the OPG/RANKL balance in human PDL fibroblasts, enhancing osteoclastogenesis (Graves et al 2020). In rat models, the increased RANKL/OPG ratio in ligature-induced periodontitis is reversed by OPG-Fc, a RANKL inhibitor. Ferroptosis, an iron-dependent form of cell death, is enhanced in diabetes, contributing to bone resorption (Zhang et al 2022). Hyperglycemia-induced ferroptosis impairs osteoblast function and worsens diabetic osteoporosis.
Beyond promoting bone resorption, diabetes inhibits bone repair by impairing osteoblast differentiation and matrix production, linked to reduced expression of Runx-2, Dlx5, and c-fos and increased NF-κB activation (Graves et al 2020; Zhou and Graves 2022). Diabetes also elevates sclerostin, advanced glycation end products (AGEs), reactive oxygen species (ROS), and TNF, suppressing Wnt signaling and bone formation (Zhou and Graves 2022). Hyperglycemia reduces IGF-1, while TNF decreases growth factors including FGF-2, TGF-β1, BMP-2, and BMP-6, limiting the proliferation of bone-lining cells (Graves et al 2020; Zhou and Graves 2022). TNF inhibitors reduce alveolar bone loss by increasing osteoblasts and reducing apoptosis, as shown by gene expression profiling (Andriankaja et al 2012; Pacios et al 2013). Apoptosis inhibition via caspase-3 inhibitors enhances bone formation (Pacios et al 2013) (Fig 4A). Diabetes also impairs mesenchymal stem cell function essential for periodontal and bone regeneration. Hyperglycemia shifts human periodontal ligament stem cell differentiation from osteogenic to adipogenic lineages, with increased adipogenic and decreased osteogenic marker expression (Graves et al 2020; Zhou and Graves 2022). AGEs, through the receptor for advanced glycation end products, increase bone loss and inflammation by disrupting coupled bone formation (Lalla and Papapanou 2011; Liu et al 2015).
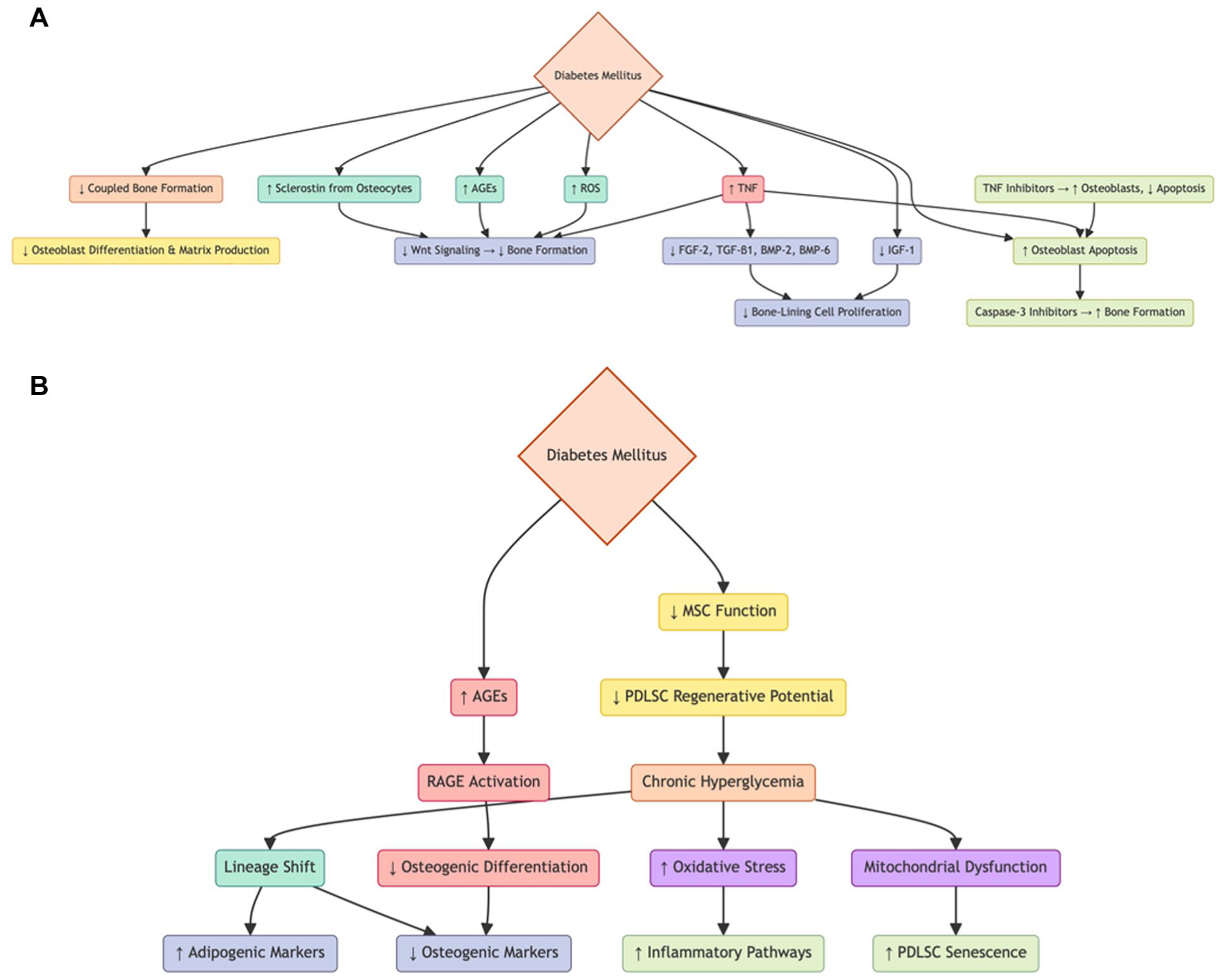
(
Oxidative Stress and Mitochondrial Dysfunction
Oxidative stress and mitochondrial dysfunction under diabetic conditions activate inflammatory pathways and promote cellular senescence in periodontal ligament stem cells (Zhou et al 2025) (Fig 4B). Other factors are oxidative stress and mitochondrial dysfunction that negatively affect the interaction between diabetes and periodontal disease (Zhou et al 2025). Hyperglycemia increases the production of ROS in periodontal tissues, overwhelming the antioxidant defense systems and resulting in oxidative damage to lipids, proteins, and mitochondrial DNA. This oxidative imbalance leads to mitochondrial membrane depolarization, adenosine triphosphate depletion, and impaired mitophagy, leading to increased NLRP3 inflammasome activation and NF-κB signaling and a proinflammatory environment (Zhou et al 2025). These processes induce cellular senescence, marked by elevated expression of p16 and p21 and the senescence-associated secretory phenotype, which contributes to local tissue destruction and impaired regeneration (Zhou et al 2025).
Mechanisms Supporting Periodontitis as a Risk Factor for Glycemic Dysregulation
The bidirectional relationship between T2DM and periodontitis may reflect the common biological pathways that often occur in the same individuals. Recent evidence suggests that they may adversely affect each other (Luong et al 2021). Current work suggests that the biological relationship between T2DM and periodontitis is based on differences in oral microbiota as well as an exaggerated inflammatory response to bacterial challenge (Graves et al 2020).
Although the biological mechanisms linking the oral microbiome and periodontitis to impaired glucose regulation are not fully understood, growing evidence implicates immune function, neutrophil activity, and cytokine biology, potentially influenced by epigenetic changes in the gingiva or during hematopoiesis (Yang et al 2024; Hajishengallis et al 2025). Chronic inflammation is central to impaired glucose tolerance and the development or worsening of T2DM via systemic microinflammation (Lalla and Papapanou 2011; Wei et al 2024). Periodontitis may increase bacteremia (reviewed in Kebschull et al 2010) and alter immune responses, enhancing both local and systemic inflammation (Graves et al 2020; Ebersole et al 2025). In the periodontium, inflammation upregulates genes related to host defense, apoptosis, and complement pathways while downregulating anti-inflammatory peroxisome proliferator-activated receptor–α (Hajishengallis et al 2025). Oral bacteremia and systemic inflammation may impair insulin signaling (Lalla and Papapanou 2011; Graves et al 2020); TNF-α can bind insulin receptors, blocking their action and promoting hyperinsulinemia (Hotamisligil 2006). Elevated cytokines (IL-10, sICAM-1, IL-1β) are linked to deeper probing depths in Hispanic adults with T2DM, while serum gingival crevicular fluid IL-α and IL-1β correlate with periodontal measures (Andriankaja et al 2023). These findings support a role for oral microinflammation in worsening T2DM (Stohr et al 2021; Oliveira et al 2023).
Microinflammation from periodontitis appears to induce systemic effects via activation in the circulation or peripheral tissues (Noz et al 2021). Activated macrophages may interact with adipocytes, leading to adipokine production that impairs glycemic control. A recent single-cell RNA analysis of peripheral blood mononuclear cells found elevated transcripts encoding proinflammatory cytokines in patients with periodontitis—both with normal glucose tolerance and with diabetes (PDDM). Notably, levels of resistin and its receptor CAP1 were elevated in periodontitis and PDDM patients, suggesting another potential link between periodontitis and DM. Resistin (“resistance to insulin”) was named for its interference with insulin action (Kamil et al 2023). In humans, resistin is a 108-amino-acid polypeptide expressed in adipocytes, pancreatic cells, muscle, and especially mononuclear cells (Kamil et al 2023). Individuals with severe insulin resistance show higher resistin levels than those with normal insulin action (Zaidi and Shirwany 2015), although several studies report no significant correlation between resistin and insulin resistance in diabetic groups (Su et al 2019). A recent meta-analysis reviewed 15 studies, using 10 with Pearson coefficients, and found a weak correlation between serum resistin and insulin resistance in obese and T2DM patients. Subgroup analysis showed this correlation only in individuals with elevated serum resistin; those with normal levels showed no relationship.
Periodontitis is thought to cause systemic effects, including impaired glycemic control, in part through bacteremia. Patients with rheumatoid arthritis and periodontitis show a dramatic increase in bacteremia (Brewer et al 2023), and a similar association may exist in diabetes. A reduced immune response to pathogens and their by-products, such as lipopolysaccharide from gram-negative bacteria, may trigger systemic inflammatory cascades resembling the proinflammatory state seen in obesity and related conditions such as metabolic syndrome, hypertension, and dyslipidemia. These conditions involve the activation of 2 key inflammatory pathways: stress-activated Jun N-terminal kinases (JNK) and NF-κB (Tsalamandris et al 2019), leading to the production of cytokines and other bioactive substances—TNF-α, IL-1, IL-6, IL-10, leptin, adiponectin, resistin, monocyte chemoattractant protein, angiotensinogen, chemokines, and serum amyloid protein—collectively called adipokines. In addition, the infiltration of adipose tissue by macrophages and immune cells (B and T cells) promotes chronic low-grade inflammation, reducing insulin sensitivity. Beyond insulin resistance, inflammatory cytokines such as IL-6 can induce pancreatic islet apoptosis, impairing insulin secretion and contributing to T2DM progression from prediabetes (Tilg and Moschen 2008). TNF-α also links insulin resistance, obesity, and islet inflammation (Tilg and Moschen 2008). In contrast, peridontitis is associated with reductions in inflammation-resolving cells (eg, T-regs, M2 macrophages) and anti-inflammatory mediators such as IL-10 (Kobayashi et al 2011; Graves et al 2020).
Epigenetic Changes Induced by Diabetes
Diabetes through high glucose and other mechanisms causes changes in histones and DNA methylation that significantly enhance the expression of proteolytic enzymes and proinflammatory mediators locally in the gingiva and periodontal tissue (Zhao et al 2023). Chronic hyperglycemia and oxidative stress in diabetes modify epigenetic regulators including DNA methyltransferases, histone acetylases/deacetylases, and miRNAs, leading to dysregulated inflammatory gene expression in periodontal tissue (Veysari et al 2023). This scoping review evaluated 30 studies and confirmed that specific epigenetic changes, including altered DNA methylation patterns and histone modifications, can modulate inflammatory responses, highlighting their potential as therapeutic targets and diagnostic markers for periodontitis (Liaw et al 2022). Diabetic periodontal tissues exhibit enhanced macrophage senescence, amplifying inflammatory responses, which can also be reversed by metformin treatment (Wang et al 2021). Epigenetic changes caused by diabetes significantly delay gingival wound healing, which is rescued by treatment with epigenetic inhibitors that block permissive histone methylation and reduce demethylation of DNA (Yang et al 2024). Diabetic conditions significantly increase histone H3 lysine 4 trimethylation (H3K4me3) and SETD1A expression in rat gingival tissues, leading to elevated levels of tissue-degrading enzymes MMP-1 and MMP-13 in human gingival fibroblasts exposed to high glucose (Kojima et al 2024). Blocking SETD1A reversed these effects, indicating a direct link between hyperglycemia-induced histone methylation changes and gingival tissue breakdown.
Epigenetic changes also occur at the systemic level by reprogramming bone marrow hematopoietic stem and progenitor cells, which may causally link comorbidities such as periodontitis with systemic conditions such as diabetes (Hajishengallis et al 2025). Periodontal infection and dysbiosis can induce epigenetic changes leading to sustained systemic inflammation and enhanced responses to subsequent inflammatory stimuli—a phenomenon known as trained immunity (Netea et al 2016). Hyperglycemia in diabetes similarly promotes central trained immunity (TRIM), causing persistent proinflammatory alterations in bone marrow progenitors that are inherited by their myeloid descendants (monocytes, neutrophils), amplifying both local periodontal inflammation and systemic metabolic dysfunction. This maladaptive immune imprinting sustains chronic inflammation, facilitating T2DM progression and further periodontal tissue destruction. Thus, diabetes and periodontitis reciprocally reinforce a systemic proinflammatory phenotype through common mechanisms involving TRIM, suggesting interventions targeting trained immunity may benefit both conditions (Fig 1).
Nitrate–Nitrite–Nitric Oxide Entersalivary Pathway
Nitric oxide (NO) is an important signaling molecule for cardiometabolic health, regulating vasodilation, glucose metabolism, and immunity (Lundberg and Weitzberg 2022). Oral microbiota contribute significantly to NO generation via the enterosalivary nitrate–nitrite–nitric oxide (NO3-NO2-NO) pathway, converting salivary nitrate (NO3) to nitrite (NO2), which is further reduced systemically into NO (Koch et al 2017; Hajishengallis et al 2022) (Fig 1). This pathway may buffer against reduced NO availability and impaired glucose regulation. Small experimental studies demonstrate beneficial effects of this microbial pathway on blood pressure and metabolism (Govoni et al 2008; Kapil et al 2013; Woessner et al 2016; Senkus and Crowe-White 2020). Findings from the ORIGINS study link microbial biomarkers indicative of increased nitrite-generation capacity with lower cardiometabolic risk scores. The effects of periodontal interventions on cardiometabolic health through microbiome alterations and the NO3-NO2-NO pathway remain to be explored, particularly given emerging evidence of both adverse and beneficial microbiome impacts on metabolic health.
Oral–Gut Axis
Gut dysbiosis is increasingly linked to cardiometabolic disease (Wang and Jia 2016), and 2 hypotheses explain the oral-to-gut microbial translocation: the hematogenous route, supported by studies of systemic bacteremia from oral sources, and the enteral route, in which swallowed oral microbiota survive gastric acidity (particularly if acidity is reduced pharmacologically) and colonize the gut (Rajasekaran et al 2024). Diabetes-associated gut dysbiosis increases serum uric acid, evident by elevated uric acid levels observed in periodontitis patients and greater alveolar bone loss in hyperuricemic mice with ligature-induced periodontitis (Sato et al 2021; Yamazaki and Kamada 2024). Allopurinol, a uric acid–lowering xanthine oxidase inhibitor, mitigates alveolar bone loss, highlighting uric acid’s mechanistic role (Sato et al 2021). In addition, poor glycemic control (HbA1c ≥ 8%) reduces subgingival butyrate-producing bacteria compared with better-controlled individuals (HbA1c < 7.8%) (Longo et al 2018), resulting from hyperglycemia-induced inflammation and oxidative stress. Reduced butyrate disrupts mucosal integrity and immune regulation, promoting further dysbiosis. Diabetes also elevates succinate levels by impairing mitochondrial metabolism and succinate dehydrogenase activity, leading to intracellular and extracellular succinate accumulation (Guo et al 2022). Excess succinate binds SUCNR1 receptors, promoting inflammation and dysbiosis, enhancing pathogen growth (eg, Fusobacterium nucleatum, Porphyromonas gingivalis), increasing virulence gene expression and diminishing immune tolerance and epithelial integrity (Guo et al 2022).
Conclusions
This review underscores the mechanistically complex, bidirectional relationship between diabetes and periodontitis, in which each condition exacerbates the other through shared inflammatory, microbial, and immune pathways. Diabetes impairs periodontal health, while periodontitis contributes to systemic inflammation, insulin resistance, and impaired insulin secretion, potentially increasing cardiovascular risk. Although evidence supporting diabetes as a risk factor for periodontitis relies primarily on observational and animal studies, clinical trials demonstrate that periodontal therapy modestly improves glycemic control, particularly in T2DM. Emerging observational studies link periodontitis-related oral dysbiosis to insulin resistance and prediabetes, suggesting oral health interventions may aid diabetes prevention, although supporting randomized trials are lacking. Recent mechanistic insights highlight hyperglycemia-induced microbiome alterations, complement dysregulation, nitrate reduction, and disruption of the oral–gut microbial axis as novel pathways driving this relationship. Integrating periodontal care—such as oral hygiene, scaling and root planing, and/or possibly antibiotics in select settings—into diabetes management can reduce systemic inflammation, improve insulin sensitivity, and enhance overall metabolic control, warranting further research into targeted preventive interventions.
Author Contributions
D.T. Graves, M.A. Levine, S. Aldosary, contributed to conception and design, data acquisition, analysis, and interpretation, drafted the manuscript; R.T. Demmer, contributed to data acquisition and interpretation, drafted the manuscript. All authors gave their final approval and agreed to be accountable for all aspects of the work.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institute of Dental and Craniofacial Research R01DE021921 and R01DE017732.