
Abstract
Cisplatin-based chemotherapy is a cornerstone treatment for advanced recurrent head and neck squamous cell carcinoma (HNSCC). However, the effectiveness of the treatment is often hindered by intrinsic and acquired resistance and associated toxicity, highlighting a pressing unmet clinical need. Here, our compound screening identified Aurora kinase inhibitors, particularly those targeting Aurora A kinase, as potential agents to sensitize resistant HNSCC cells to cisplatin. While Aurora kinases are well-established regulators of mitosis, their precise role in cisplatin resistance is largely unknown, given that cisplatin confers toxicity primarily in cells undergoing DNA replication. We confirmed that depletion of Aurora A or its activators enhanced cisplatin response in resistant HNSCC cells. Analyses of a comprehensive database and locally treated HNSCC patient samples revealed compelling associations between Aurora A overexpression/activation and cisplatin resistance, tumor recurrence, and poor patient survival. Pharmacologic inhibition of Aurora A effectively synergized with cisplatin treatment in cellular assays and a syngeneic mouse tumor model of HNSCC. Mechanistically, Aurora A inhibition enhanced apoptosis induction after cisplatin treatment, particularly in S-phase cells; induced replication stress; and suppressed the repair of cisplatin-induced DNA crosslinking. Taken together, our findings shed light on important functions of Aurora A kinase beyond mitotic regulation. The multifaceted roles of Aurora A suggest its potential as a prime anticancer drug target. Given the ongoing investigations into numerous Aurora inhibitors for cancer therapy, exploring their clinical applications in HNSCC, especially in combination with platinum drugs, may hold significant promise.
This is a visual representation of the abstract.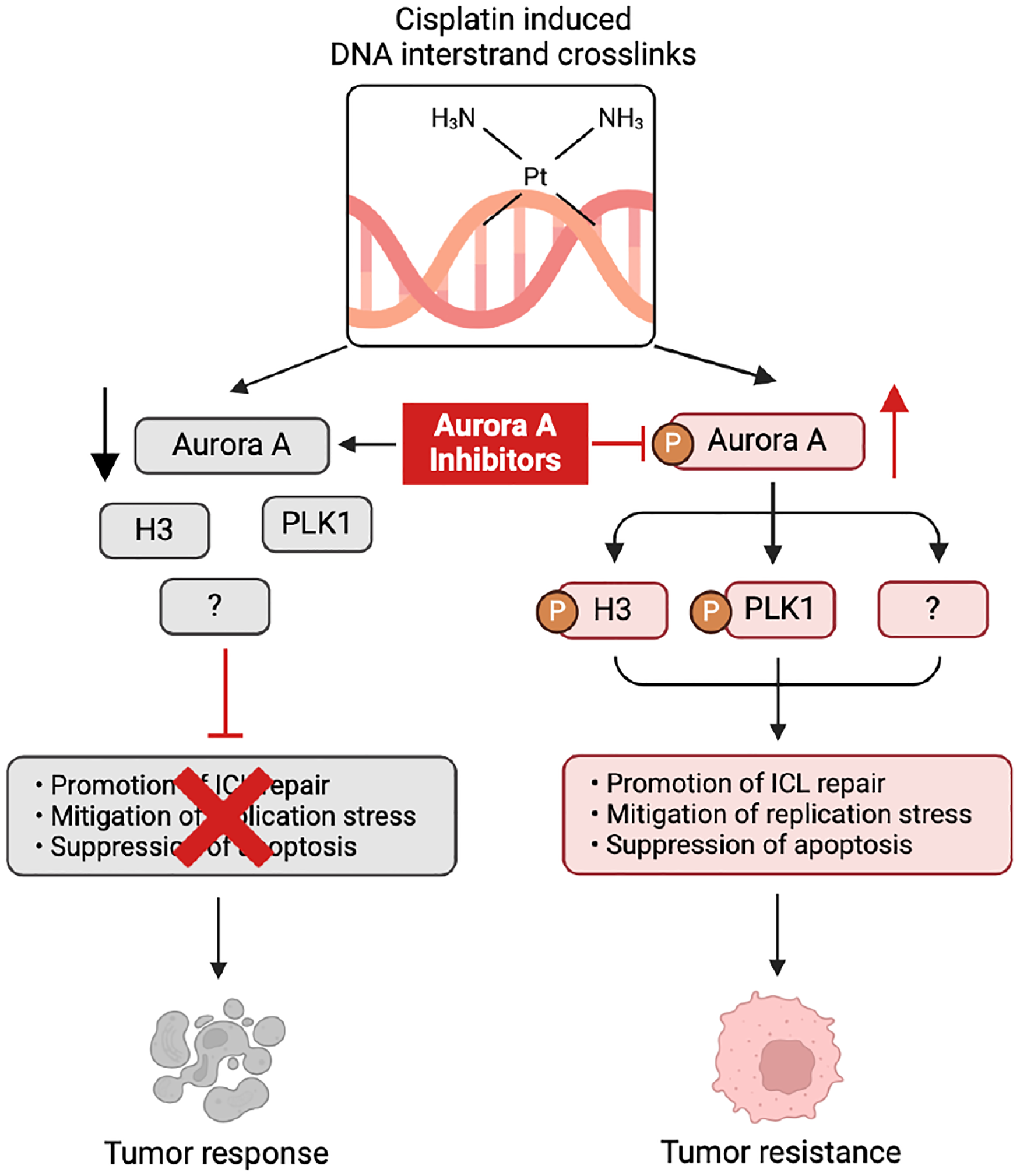
Keywords
Introduction
Head and neck cancers are among the most prevalent cancers globally, ranking the sixth most common worldwide (Bray et al. 2024). In the United States, head and neck cancers are projected to account for 3% of all new cancer cases and 2% of cancer-related deaths in 2024, with incidence and mortality rates still rising (Siegel et al. 2024). Head and neck squamous cell carcinoma (HNSCC) accounts for 90% of head and neck cancers and is typically treated with surgery, radiation, and chemotherapy based on disease stage (Johnson et al. 2020). Cisplatin (cis-diammine-dichloro-platinum II) is used as a first-line chemotherapy treatment for patients diagnosed with HNSCC at advanced stages, to prevent tumor recurrence and improve treatment outcome (Pfister et al. 2020). Upon entering the cell, cisplatin loses its chloride ligands, allowing it to bind DNA and form DNA interstrand crosslinks (ICLs). ICL induction alters DNA structure, prevents DNA replication and other metabolism, and ultimately inhibits cell growth and induces cell death (Brown et al. 2019). Unfortunately, some tumors either exhibit limited responsiveness to cisplatin or become nonresponsive after the initial response (Kanno et al. 2021). Cisplatin resistance limits its effectiveness in HNSCC and other cancers, highlighting the pressing need to understand its mechanism and develop strategies to overcome it.
Extensive studies have revealed interesting insights into the underlying mechanisms of cisplatin resistance. For instance, increased DNA damage repair, enhanced drug efflux, and inhibition of apoptosis have been implicated as causes of treatment resistance (Kanno et al. 2021). Many strategies have been proposed to counter cisplatin resistance. For example, combining cisplatin with radiation and other chemotherapeutic agents that induce different types of DNA damage may reduce resistance in tumor cells (Kiss et al. 2021). Additional treatment combinations have also been suggested, involving the growth factor receptor (EGFR) monoclonal antibody that is currently used in combination with radiation in human papillomavirus–negative HNSCC (Fasano et al. 2021), and the programmed cell death 1 receptor (PD-1) and its ligand programmed cell death ligand 1 (PD-L1) immune checkpoint inhibitors that have joined the primary treatment options for recurrent or metastatic HNSCC (Mei et al. 2020). Furthermore, targeted therapies that disrupt cisplatin-induced DNA damage repair or promote cell death have been sought in combination with cisplatin to confer a more effective and selective cytotoxic effect, as cancer cells often evolve dysregulation of specific mechanisms to evade cisplatin-induced cytotoxicity (Curtin 2012; Rocha et al. 2018).
Aurora kinases, a family of serine/threonine kinases, comprise Aurora kinase A (AURKA), B (AURKB), and C (AURKC). Both Aurora kinase A and B are essential for cell division, playing key roles in different aspects of mitotic progression (Willems et al. 2018). Aurora kinase A is crucial for centrosome maturation, spindle assembly, and accurate chromosome segregation during mitosis. It ensures proper cell-cycle progression from the G2 phase to mitosis, helping to maintain chromosomal stability (Willems et al. 2018). Aurora B is involved in chromosome condensation, alignment, and segregation as well as spindle assembly checkpoint and cytokinesis (Willems et al. 2018). Interestingly, Aurora kinases have attracted extensive interest in cancer research. Overexpression or hyperactivation of Aurora kinases is commonly observed in various types of cancer, and targeting Aurora kinases has emerged as a new strategy to disrupt tumor growth (Du et al. 2021). Unfortunately, despite the pharmaceutical development and numerous clinical trials of selective Aurora inhibitors, none has obtained U.S. Food and Drug Administration approval, suggesting that suppressing Aurora kinases to hamper cell proliferation may not yield a therapeutic window necessary for cancer treatment. On the other hand, one may posit that the discovery of synergistic drug combinations and characterization of prognostic makers hold the key to the ultimate clinical success of Aurora inhibition.
In HNSCC, the overexpression of both Aurora A and Aurora B was shown to be associated with cancer progression and with adverse metastasis-free survival or overall survival (Tanaka et al. 2005; Pannone et al. 2011). The efficacy of Aurora inhibition was illustrated in many preclinical studies. For example, HNSCC cells with mutant p53 were found to be sensitive to Aurora inhibition monotherapy (Marxer et al. 2014), an inhibitor of WEE1 synergized with Aurora A inhibitors in a HNSCC xenograft tumor model in mice (Lee et al. 2019); Aurora suppression overcame HNSCC resistance to EGFR inhibitors (Hoellein et al. 2011). However, despite the promising preclinical findings, clinical trials assessing the efficacy of Aurora kinase inhibitors, either alone or in conjunction with radiation, EGFR inhibitors, or PD-1 inhibitors, have been very limited. Thus far, these trials have demonstrated only modest success in managing the disease or eliciting partial responses in minor subsets of patient populations. Here, we identified inhibitors of Aurora A kinase as effective agents that overcame cisplatin resistance in both HNSCC cells and tumors. Our investigations revealed a significant role of Aurora A in mediating cell survival following cisplatin treatment, with its inhibition leading to reduced DNA repair and increased replication stress and apoptosis. These findings present compelling evidence supporting the targeting of Aurora A as a viable strategy in cisplatin-based therapy for HNSCC.
Materials and Methods
Detailed materials and experimental procedures are provided in the appendix. The study complied with ARRIVE 2.0 guidelines. Procedures of all the animal experiments were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Results
Characterization of Aurora Kinases as Effective Targets to Enhance Cisplatin Response in HNSCC
We performed unbiased screening of 463 small-molecule inhibitors of protein kinases and epigenetic regulators and identified several inhibitors of Aurora kinases, especially selective inhibitors of Aurora A, as top candidates that significantly enhanced the effect of cisplatin (Fig. 1A). These inhibitors, in combination with cisplatin, effectively suppressed cell viability in SCC11B, a cell line highly resistant to cisplatin (Wang et al. 2012) (Fig. 1A). We sought to confirm these findings using small interfering RNA (siRNA) to deplete Aurora A, Aurora B, or interacting activators of Aurora A, namely, Ajuba, TPX2 (target protein for Xenopus kinesin-like protein 2), and Bora (Aurora borealis) (Carmena et al. 2009) (Fig. 1B, C). In SCC11B and SCC38, 2 cisplatin-resistant HNSCC cell lines, depletion of Aurora kinases or Aurora activators substantially enhanced the cisplatin response, measured by suppression of cell viability (Fig. 1D–H). We then conducted siRNA-mediated Aurora A knockdown in murine SCC-VII cancer cells (Appendix Fig. 1A) and also observed significantly increased cisplatin sensitivity (Appendix Fig. 1B). In contrast, the depletion of Aurora kinases did not markedly alter the susceptibility to cisplatin in HaCaT, a nontumorigenic human keratinocyte cell line (Fig. 1I). Thus, Aurora kinases, particularly Aurora A, may act as a pivotal mediator of cisplatin resistance, and their suppression confers effective, and potentially selective, cisplatin sensitization in HNSCC.
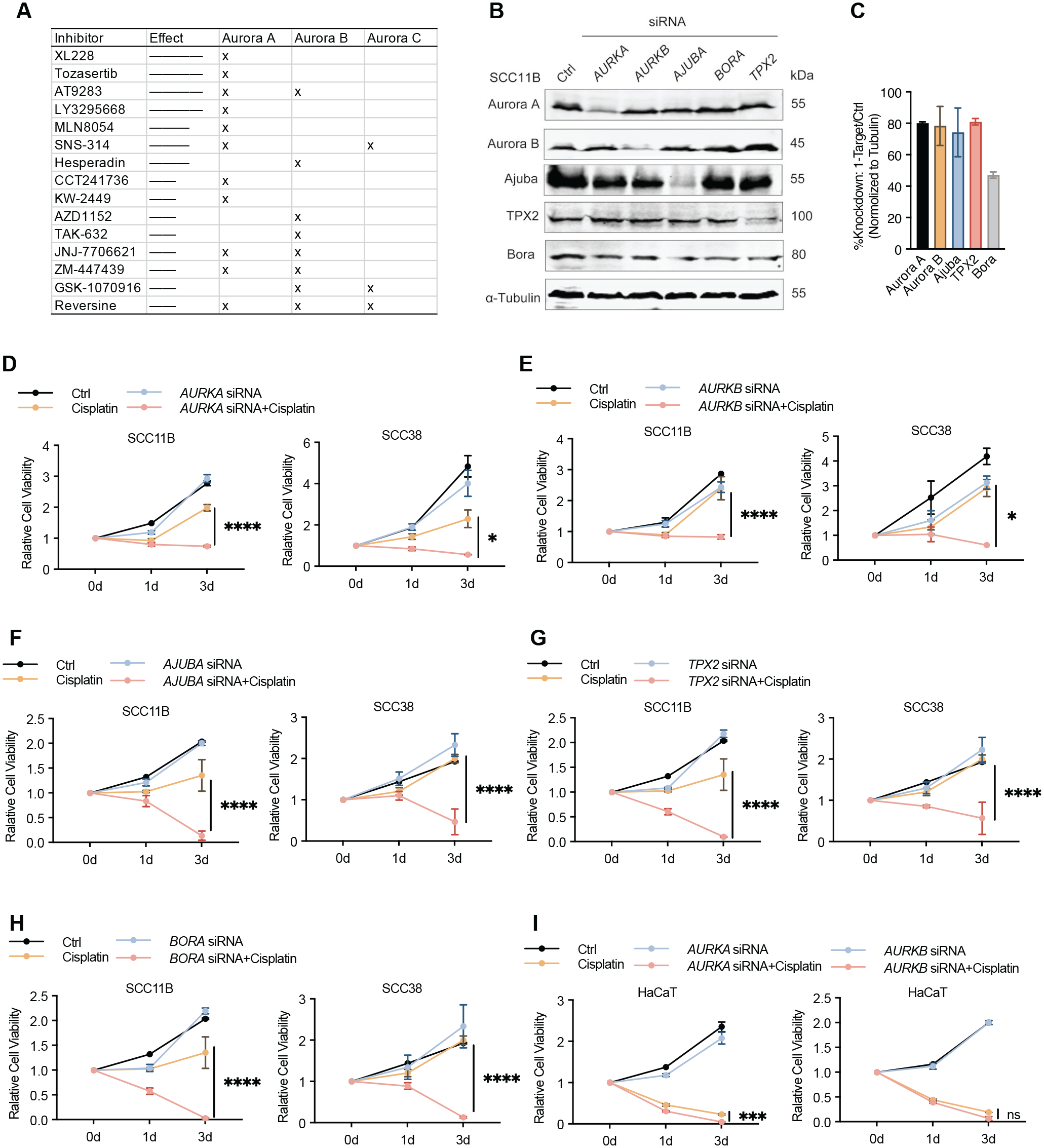
Characterization of Aurora kinases as effective targets to enhance cisplatin response in head and neck squamous cell carcinoma. (
Expression Level and Kinase Activity of Aurora A Are Associated with Tumor Resistance and Recurrence in HNSCC
To explore the clinical relationship between Aurora A and the prognosis of HNSCC, we analyzed the transcriptome data from the TCGA Head and Neck Cancer dataset. Elevated Aurora A expression was associated with poor overall survival in HNSCC (Appendix Fig. 2A). Although similar trends were observed for Aurora A activators Ajuba, TPX2, and Bora, the associations between their expression and HNSCC mortality were much less significant (Appendix Fig. 2B–D). In comparison, the expression of Aurora B and Aurora C kinases was not associated with poor survival (Appendix Fig. 2E, F).
Interestingly, the association between Aurora A expression and HNSCC mortality was seen exclusively in patients with stage III and IV HNSCC but not those with stage I and II HNSCC (Fig. 2A, B). Moreover, the strongest elevation of Aurora A expression was seen in stage IV HNSCC patients (Fig. 2C). Of various primary treatment outcomes, the group with persistent disease exhibited the highest level of Aurora A expression, whereas that with complete response harbored the lowest Aurora A expression (Fig. 2D). Aurora A expression also positively correlated with tumor recurrence after the initial treatment (Fig. 2E).
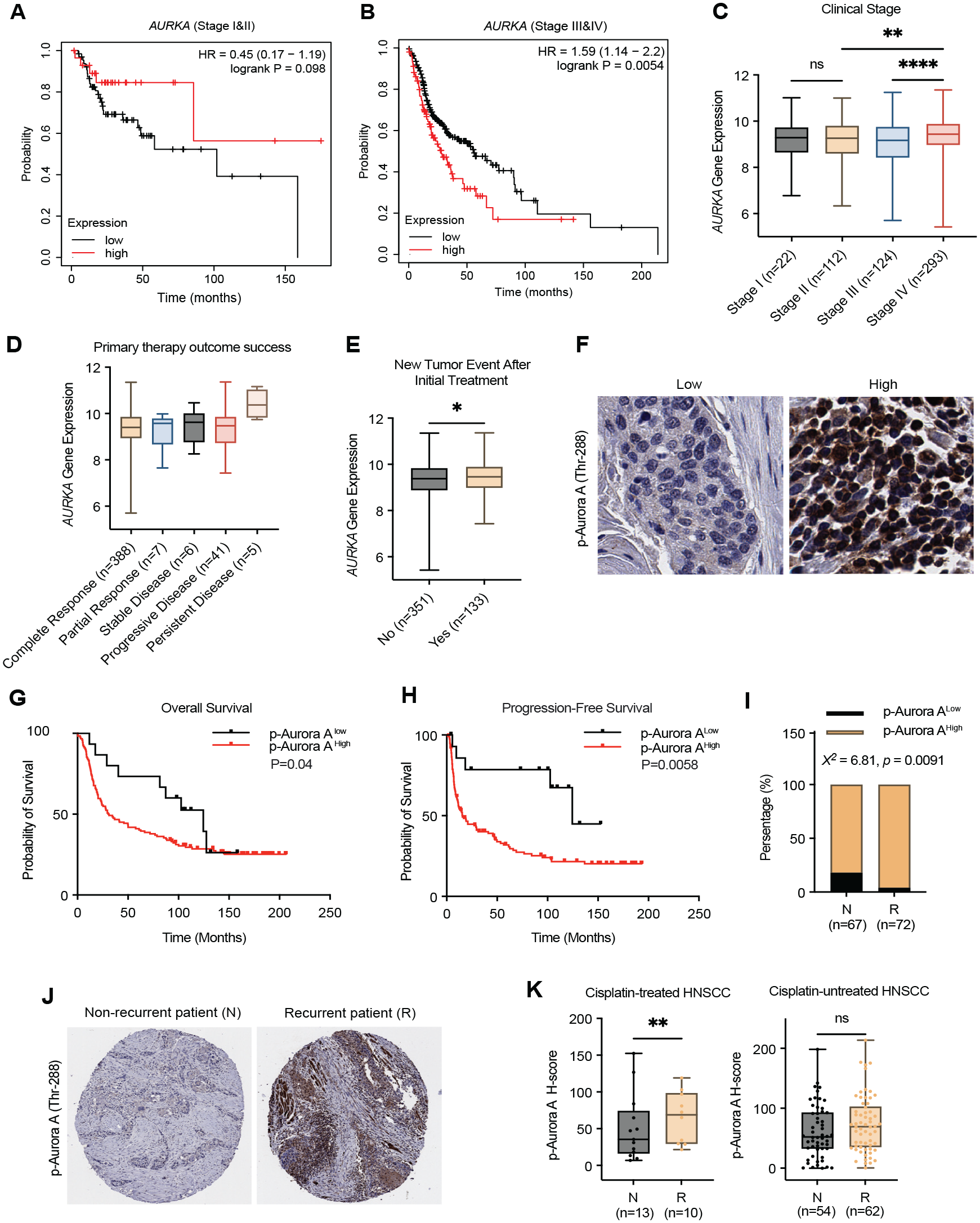
The expression level and kinase activity of Aurora A are associated with tumor resistance and recurrence in head and neck squamous cell carcinoma (HNSCC). (
We then asked if Aurora A kinase activation was associated with the treatment outcome and survival by analyzing 139 HNSCC patient tumor samples using immunohistochemistry (IHC) for Aurora A phosphorylation at Thr-288, an indicator of its activation (Ohashi et al. 2006) (Fig. 2F). The Kaplan–Meier survival analysis revealed a significant correlation between Aurora A activation and adverse overall survival or progression-free survival (Fig. 2G, H). Stratifying the patient cohort by stage revealed that Aurora A kinase activity was strongly associated with poor survival outcomes in stage III/IV patients, whereas this association was significantly weaker in stage I/II patients (Appendix Fig. 2G, H). The different association of Aurora A kinase activity with patient survival in stage I/II and stage III/IV is potentially due to the distinct treatment options (e.g., surgery vs. chemoradiation). The levels of Aurora A activation were significantly higher in tissues of recurrent HNSCC patients, compared with those with nonrecurrent HNSCC (Fig. 2I, J). Finally, we separated these samples into 2 cohorts with or without cisplatin treatment. Strikingly, Aurora A activation, quantified by H-score of IHC analysis, correlated with tumor recurrence in HNSCC treated with adjuvant cisplatin but not in cisplatin-untreated HNSCC (Fig. 2K). Together, our findings established Aurora A expression and activation as markers of HNSCC prognosis, particularly for cisplatin resistance, tumor recurrence, and patient mortality in advance HNSCC.
Pharmacologic Inhibition of Aurora A Overcomes Cisplatin Resistance in HNSCC
Building on the results of our compound screening and patient sample analyses, we sought to validate 3 Aurora A inhibitors identified as top hits in our drug screen assay, XL228, Tozasertib (VX-680), and AT9283, in cisplatin-based HNSCC treatment. In both SCC11B and SCC38 cells, combined treatment of cisplatin with XL228 (Fig. 3A), Tozasertib (Fig. 3B), and AT9283 (Appendix Fig. 3A) additively suppressed cell viability compared with cisplatin or Aurora inhibitor alone. Aurora inhibition alone induced strong cytotoxicity in certain cases, such as in SCC11B cells treated with Tozasertib or AT9283 (Fig. 3B and Appendix Fig. 3A). A less significant effect was observed in HaCaT cells (Appendix Fig. 3A, B), consistent with our earlier observation of Aurora A depletion. Replacing cisplatin with other clinical platinum drugs, oxaliplatin and carboplatin, showed similar levels of enhancement by XL228 (Fig. 3C and Appendix Fig. 3C).
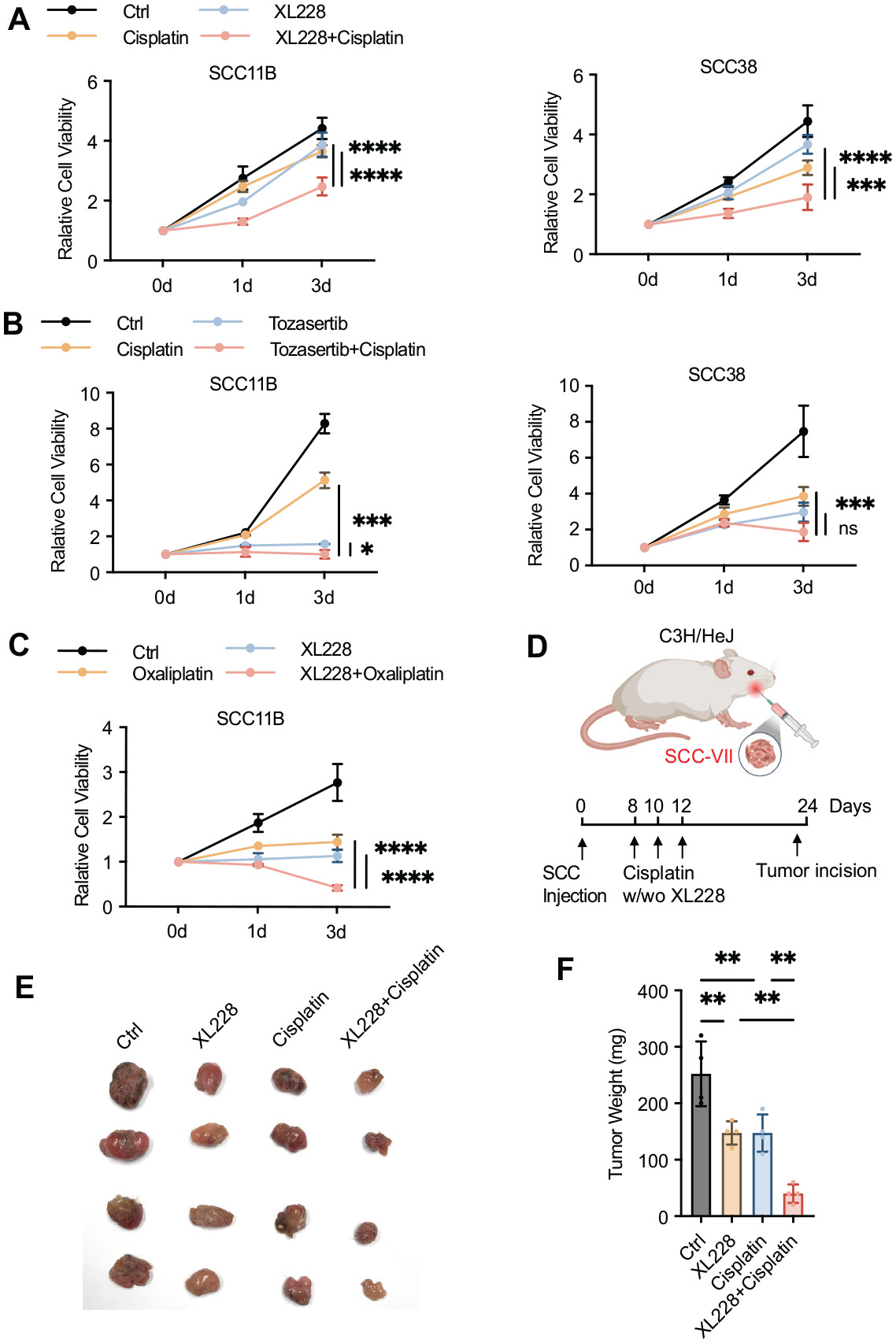
Pharmacologic inhibition of Aurora A overcomes cisplatin resistance in head and neck squamous cell carcinoma. (
We used an SCC-VII syngeneic oral squamous cell carcinoma tumor model to evaluate Aurora A inhibition in vivo, as it successfully recapitulates oral tumor growth, local invasion, and distinct metastasis, making it a valuable tool for studying HNSCC (O’Malley et al. 1997). Implantation of SCC-VII resulted in tumor formation in the oral cavity of mice, and 3 doses of cisplatin with or without XL228 were administered subcutaneously near the tumor sites (Fig. 3D). While cisplatin treatment and XL228, at their given doses, had moderate effects on SCC-VII tumors, the combined treatment exhibited a markedly enhanced outcome (Fig. 3E, F). Together, our results from the cellular assays and mouse tumor model supported the potential of Aurora A inhibition in overcoming cisplatin resistance in HNSCC.
Aurora A Inhibition Potentiates the Apoptotic Response to Cisplatin in HNSCC
We next attempted to elucidate the underlying mechanism for the role of Aurora A in mediating cisplatin resistance in HNSCC. We observed a profound impact of Aurora A inhibition on the induction of apoptosis after cisplatin treatment, with AT9283 alone moderately inducing apoptosis, detected by Annexin V staining; AT9283 combined with a sublethal cisplatin drastically increased apoptotic induction (Fig. 4A, B). We then confirmed these findings using cleaved caspase-3 as a biochemical marker of apoptosis. Interestingly, while cisplatin induced significant DNA damage, as indicated by a common DNA double-strand break marker γ-H2AX, its effect on apoptotic induction was relatively limited (Fig. 4C–E). Conversely, Aurora A inhibitors, XL228 (Fig. 4C), Tozasertib (Fig. 4D), and AT9283 (Fig. 4E), caused more significant levels of apoptosis but lower DNA damage compared with cisplatin. The combination of cisplatin and Aurora inhibition strongly elevated the levels of DNA damage and apoptosis (Fig. 4C–E). This synergistic effect in apoptotic induction was also observed in murine SCC-VII cells treated with XL228 (Appendix Fig. 4A) or AT9283 (Appendix Fig. 4B), in combination with cisplatin.
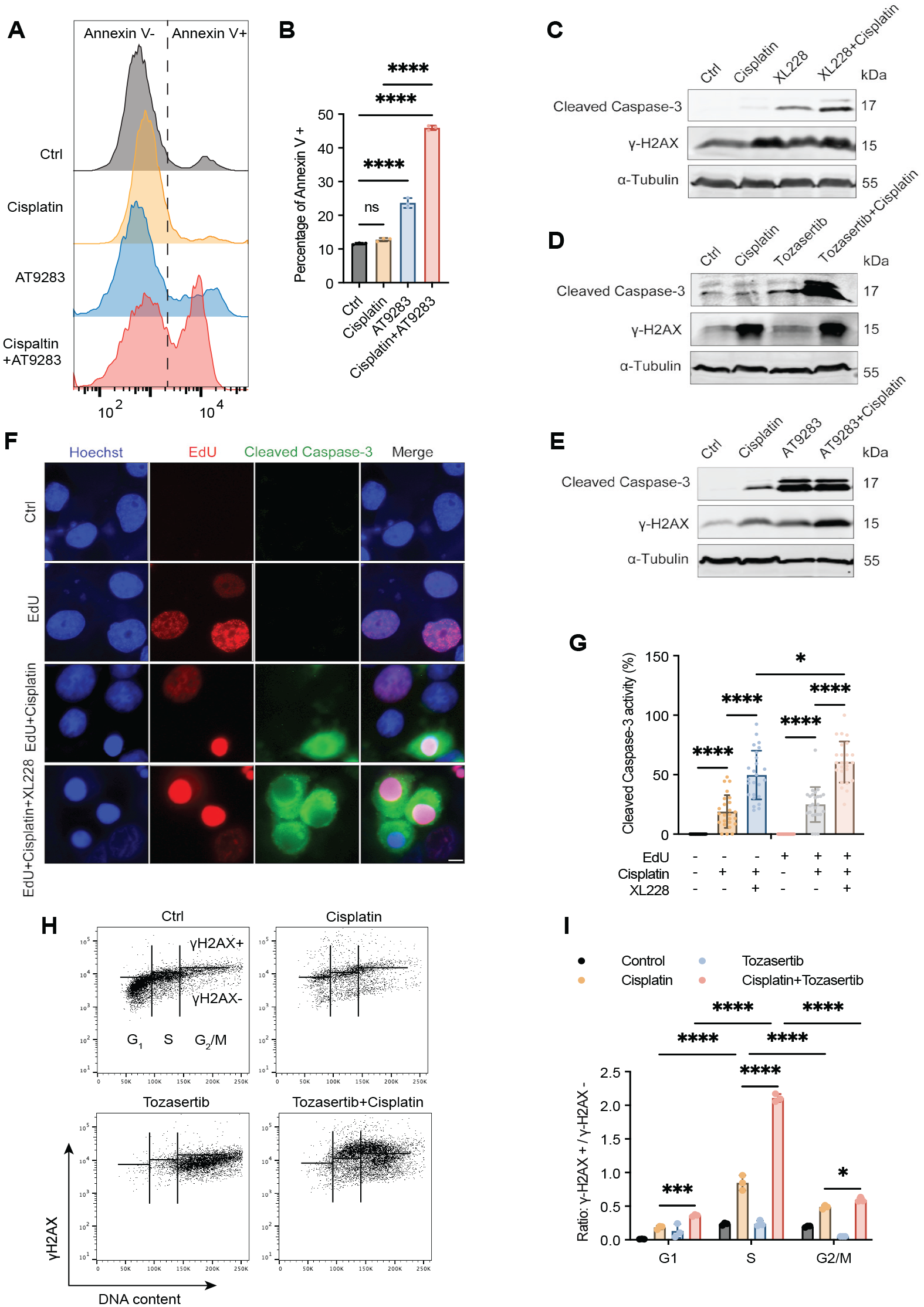
Aurora A inhibition potentiates the apoptotic response to cisplatin in head and neck squamous cell carcinoma. (
Cisplatin is known to disrupt DNA replication as a main cause of cytotoxicity. On the other hand, Aurora A functions as a mitotic kinase to promote mitotic entry and progression. Thus, we further investigated the cisplatin and Aurora A inhibition combination in the context of the cell cycle. Cisplatin and XL228 treatment led to cell-cycle arrest in the S and G2/M phases, respectively, as expected, and their combination exhibited cell-cycle arrest in both the S-phase and, to a lesser extent, G2/M (Appendix Fig. 4C, D). We used 5-ethynyl-2′-deoxyuridine (EdU) to label newly synthesized DNA and mark cells in the S-phase. Notably, XL228 sensitized caspase-3 cleavage in both EdU-negative and -positive cells, whereas the synergistic effect of XL228 and cisplatin was more profound in EdU-positive cells compared with EdU-negative cells (Fig. 4F, G). Furthermore, we used flow cytometry–based analysis of γ-H2AX to reveal the cell-cycle dependence of DNA damage induction. Interestingly, Aurora A inhibition strongly augmented cisplatin-induced DNA damage, predominant in S-phase cells (Fig. 4H, I). A moderate increase in γ-H2AX was observed in the G2/M phase, potentially reflecting the role of Aurora A kinase in the G2/M transition or due to cell-cycle progression from the S phase to G2/M with unresolved replication stress (Fig. 4H, I). Thus, our findings established the role of Aurora A in suppressing cisplatin-induced apoptosis, largely attributed to its functions in the S-phase of the cell cycle.
Aurora A Inhibition Induces Replication Stress and Suppresses the Repair of Cisplatin-Induced DNA Crosslinking
All 3 Aurora A inhibitors, XL228, Tozasertib, and AT9283, increased γ-H2AX and caspase-3 cleavage, along with reduced histone H3 Ser-10 phosphorylation, a biochemical marker of mitosis (Fig. 5A–C). Interestingly, these inhibitors also elevated the levels of replication protein A (RPA) phosphorylation (Fig. 5A–C). Phosphorylation of RPA at the N-terminus of its 32-kDa subunit (RPA32) is induced upon replication stress, facilitating DNA replication fork protection, repair, and DNA damage checkpoint signaling, with its posttranslational modifications serving as well-established markers of replication stress (Maréchal and Zou 2014; Byrne and Oakley 2019). Depletion of Aurora A, or that of Aurora B, Ajuba, TPX2, and Bora, induced RPA32 phosphorylation, with Aurora A and TPX2 being the more effective targets among them (Fig. 5D). We then analyzed RPA32 phosphorylation in both whole-cell lysates and chromatin fractions, enriched using a well-established fractionation protocol (Fig. 5E). Chromatin isolated from S-phase cells exhibited a higher basal level of RPA phosphorylation compared with G1-phase (Fig. 5F). RPA32 phosphorylation was increased upon treatment with cisplatin or Aurora inhibitors, while the combination of them further increased RPA32 phosphorylation; similar effects were seen in both whole-cell lysates and chromatin fractions (Fig. 5F).
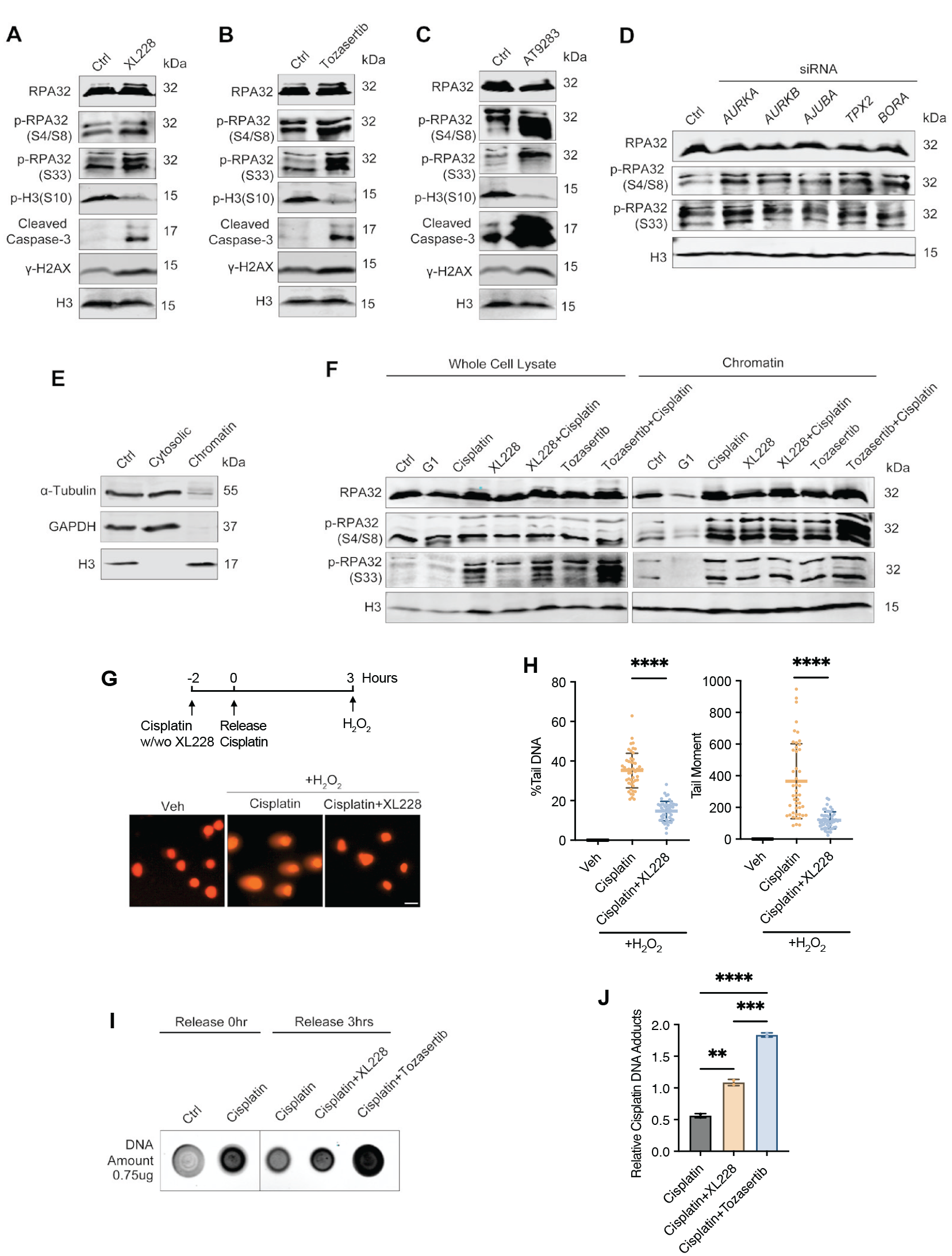
Aurora A inhibition induces replication stress and suppresses the repair of cisplatin-induced DNA crosslinking. (
We then asked if Aurora A facilitates DNA ICL repair, which is a pivotal mechanism to render cells resistant to cisplatin. A modified single-cell gel electrophoresis (comet assay) was used to assess the removal of ICLs (Fig. 5G). Interestingly, XL228 treatment substantially impeded the removal of ICLs, as quantified by both percentages of DNA in the tail and tail moment (Fig. 5G, H). We then confirmed the finding by dot blotting of genomic DNA using an anti-cisplatin DNA adducts antibody. Both XL228 and Tozasertib significantly reduced the efficiency of ICL repair, resulting in higher levels of residential cisplatin-DNA adducts (Fig. 5I, J).
Discussion
In this study, we identified inhibitors of Aurora A kinase as potentially promising therapeutics that substantially improved the treatment outcome of cisplatin in resistant HNSCC cells and tumors. Our functional studies further demonstrated the role of Aurora A kinase in facilitating ICL repair, preventing replication stress, and antagonizing the induction of cell death. These findings are in line with the clinical association of Aurora A expression and kinase activation with HNSCC aggressiveness and treatment resistance. The findings hold strong and broad implications for cancer therapy as cisplatin is used in half of chemotherapeutic regimens across all types of cancer.
Aurora A is a well-established regulator of mitotic progression (Willems et al. 2018). Here, our studies suggested important functions of Aurora A beyond mitotic progression, particularly in S-phase. Aurora A inhibition or depletion caused replication stress, indicated by RPA32 phosphorylation. Inhibition of Aurora A strongly enhanced the induction of DNA damage and apoptosis in S-phase cells upon cisplatin treatment. Aurora A was also required for the efficient removal of DNA ICLs. These functions of Aurora A are in good harmony with the efficacy of Aurora A inhibitors in cisplatin sensitization. However, future investigations are needed to reveal specific downstream pathways responsible for these functions of Aurora A. Interestingly, Plk1, an established substrate of Aurora A in the G2/M transition (Joukov and De Nicolo 2018), was also shown to promote replication stress responses. By phosphorylating DNA replication factors, including origin recognition complex 2 (ORC2) and Primpol, PLK1 plays roles in coordinating replication initiation and elongation and mediating the protection and recovery of stalled replication forks (Song et al. 2011; Song et al. 2012; Bailey et al. 2021). Potentially aligned with our findings, PLK1 suppression also led to cisplatin sensitization in esophageal squamous cell carcinoma, lung adenocarcinoma, and HNSCC (Wu et al. 2019; Hagege et al. 2021; Wang et al. 2023). Furthermore, despite being commonly regarded as a mitotic marker, histone H3 Ser-10 phosphorylation has been implicated in S-phase, with a potential role in the replication stress response (Chen et al. 2018). Aurora A was also shown to phosphorylate geminin to govern the proper prereplication complex formation (Tsunematsu et al. 2013). Nevertheless, further elucidation is required regarding additional substrates and downstream mechanisms to fully comprehend the roles of Aurora A in the replication stress response, apoptotic regulation, and DNA ICL repair, as reported in this study.
Numerous inhibitors of Aurora A and Aurora B have been evaluated in clinical trials; however, the clinical success of Aurora kinase inhibition remains to be demonstrated, potentially with the development of more selective and potent inhibitors and, importantly, with mechanism-based identification of effective treatment combinations and prognostic biomarkers. Our findings particularly support the use of Aurora A inhibitors in combination with cisplatin or similar platinum drugs. We validated the efficacy of these combinations in HNSCC cells and the SCC-VII tumor model. The use of Aurora A inhibitors to overcome cisplatin resistance has been previously shown in human lung cancer A549 cells (Xu et al. 2014), esophageal adenocarcinoma OE33 cells and tumor xenografts (Sehdev et al. 2012), and ovarian cancer OVCAR-3 cells and tumor xenografts (Winardi et al. 2022). In HNSCC, studies have shown that high expression of Aurora A correlated with disease progression and decreased overall survival rates (Li and Zhang 2015; Dawei et al. 2018). Our analysis of the HNSCC database supported the prognostic value of Aurora A expression, especially in patients with advanced HNSCC. Using locally treated HNSCC samples, we demonstrated that Aurora A kinase activation can also serve as a biomarker predictive of cisplatin resistance, tumor recurrence, and poor patient mortality in advanced HNSCC. Thus, we believe that HNSCC patients in advanced stages, harboring elevated levels of Aurora A expression and kinase activation, are likely to benefit from the combinatorial treatment of cisplatin with Aurora A inhibition.
Authors’ Contributions
X. Li, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; Z. Wang, L. Wang, E.A. Lanzel, M.R. Buchakjian, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; G.G. Oakley, contributed to conception, design, data analysis and interpretation, critically revised the manuscript; A. Peng, contributed to conception, design, data analysis and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345241309624 – Supplemental material for Targeting Aurora A to Overcome Cisplatin Resistance in Head and Neck Cancer
Supplemental material, sj-docx-1-jdr-10.1177_00220345241309624 for Targeting Aurora A to Overcome Cisplatin Resistance in Head and Neck Cancer by X. Li, Z. Wang, G.G. Oakley, L. Wang, E.A. Lanzel, M.R. Buchakjian and A. Peng in Journal of Dental Research
Footnotes
Acknowledgements
We thank Drs. Quynh-Thu Le and Susan Knox (Stanford University) for reagents and Dr. Xian Jin Xie (University of Iowa) for stimulating discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the National Institutes of Health (CA233037, DE030427) to A. Peng.
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.