
Abstract
Due to its capacity to drive osteoclast differentiation, the receptor activator of nuclear factor kappa-β ligand (RANKL) is believed to exert a pathological influence in periodontitis. However, RANKL was initially identified as an activator of dendritic cells (DCs), expressed by T cells, and exhibits diverse effects on the immune system. Hence, it is probable that RANKL, acting as a bridge between the bone and immune systems, plays a more intricate role in periodontitis. Using ligature-induced periodontitis (LIP), rapid alveolar bone loss was detected that was later halted even though the ligature was still present. This late phase of LIP was also linked with immunosuppressive conditions in the gingiva. Further investigation revealed that the ligature prompted an immediate migration of RANK-expressing Langerhans cells (LCs) and EpCAM+ DCs, the antigen-presenting cells (APCs) of the gingival epithelium, to the lymph nodes, followed by an expansion of T regulatory (Treg) cells in the gingiva. Subsequently, the ligatured gingiva was repopulated by monocyte-derived RANK-expressing EpCAM+ DCs, while gingival epithelial cells upregulated RANKL expression. Blocking RANKL signaling with monoclonal antibodies significantly reduced the frequencies of Treg cells in the gingiva and prevented gingival immunosuppression. In addition, RANKL signaling facilitated the differentiation of LCs from bone marrow precursors. To further investigate the role of RANKL, we used K14-RANKL mice, in which RANKL is overexpressed by gingival epithelial cells. The elevated RANKL expression shifted the steady-state frequencies of LCs and EpCAM+ DCs within the epithelium, favoring LCs over EpCAM+ DCs. Following ligature placement, heightened levels of Treg cells were observed in the gingiva of K14-RANKL mice, and alveolar bone loss was significantly reduced. These findings suggest that RANKL-RANK interactions between gingival epithelial cells and APCs are crucial for suppressing gingival inflammation, highlighting a protective immunological role for RANKL in periodontitis that was overlooked due to its osteoclastogenic activity.
Introduction
Gingival homeostasis is delicate, and disruption of the epithelial integrity might enable microbial invasion, resulting in the development of destructive inflammation termed periodontitis (Moutsopoulos and Konkel 2018). This pathologic process is experimentally mimicked by ligature-induced periodontitis (LIP), in which the ligature exerts both mechanical and microbial challenges on the epithelium that induce rapid bone loss (Abe and Hajishengallis 2013). To coexist with the dental biofilm and to maintain epithelial integrity, the gingival epithelium contains Langerhans cells (LCs), a specialized type of dendritic cells (DCs) found exclusively in stratified epithelia (Capucha et al. 2018; Hovav 2018; Brand et al. 2023). LCs play a protective role in periodontitis induced by the pathobiont Porphyromonas gingivalis, a model in which the kinetics of alveolar bone loss are slower compared with LIP (Arizon et al. 2012). It is unknown, however, which role LCs play during LIP. Furthermore, while the immunological mechanisms underlying LIP-induced bone loss have been investigated (Lin et al. 2021), it is unclear how the gingiva copes with the chronicity of this challenge to prevent excessive damage.
In periodontitis, the receptor activator of NF-κB ligand (RANKL) and its receptor RANK are known to play a pathological role (Settem et al. 2021; Zhou and Graves 2022). Nevertheless, RANKL and RANK were initially identified as capable of controlling DC function (Anderson et al. 1997). The RANKL-RANK signaling was also shown to shape DC activity in barrier tissues. In the skin, RANKL expressed by inflamed epidermal keratinocytes interacts with RANK on LCs, resulting in the expansion of T regulatory (Treg) cells that decrease skin inflammation (Loser et al. 2006). On the other hand, during herpes simplex virus type 1 infection, the epidermal RANKL increased the anti-viral CD8+ T-cell response by protecting the LCs from apoptosis (Klenner et al. 2015). This suggests that the barrier epithelium and local DCs interact via RANKL-RANK signaling to orchestrate local immunity. Since the epidermis and the oral epithelium share similar immunological features (Hovav 2018; Brand et al. 2023), it is likely that RANKL could also play an immunological role in gingival inflammatory conditions such as periodontitis. Nevertheless, whether RANKL is expressed by the inflamed gingival epithelium and how it affects periodontal inflammation remain unknown.
Materials and Methods
Extensive experimental details are presented in the Appendix.
Animal protocols were approved by the Hebrew University Institutional Animal Care and Use Committee. The study conforms to the ARRIVE guidelines.
Results
LIP Induces Rapid Bone Loss followed by Local Immunosuppressive Conditions
To investigate the pathological progression of LIP, which induces rapid periodontal destruction (Marchesan et al. 2018), we quantified the kinetics of alveolar bone loss. Significant bone loss was detected 3 d after ligature placement, and the bone loss increased after 9 d (Fig. 1A, B). On day 21, no further bone loss was detected compared with the ninth day, even though the ligature was still present. The presence of the ligature was necessary for the bone loss, as removal of the ligature on the third day decreased the levels of bone loss (Fig. 1A, B). Since LIP-related bone loss is an immune-mediated process, inhibition of the rapid bone loss after 9 d suggests a change in gingival immunity. To examine this, we profiled the gene expression of gingival epithelial cells by RNA sequencing (RNAseq). To enable effective comparison, epithelial tissues were collected from naïve mice, mice that underwent LIP for 9 d (Ligature), and a similar group except that the ligation was removed on the third day of LIP (Removal). As depicted in Figure 1C, the hierarchical cluster analysis indicated differential gene expression patterns between the 3 experimental groups. Principal component analysis further displayed the distribution of the 3 groups based on the differential gene expression (Fig. 1D). To further understand the gene expression profile in each group, we performed gene set enrichment analysis. Comparing the Ligature and Naïve groups revealed upregulated pathways associated with tissue/epithelial repair and regeneration in the Ligature group (Fig. 1E). For instance, the E2F target pathway is essential to epidermal wound repair (D’Souza et al. 2002), while the mitotic spindle and G2/M pathways control the polarity and intercellular forces of epithelial cells, respectively (Nakajima 2018; Donker et al. 2022). The upregulation of Wnt/β-catenin signaling is also known to affect gingival regeneration (Qi et al. 2020). Yet, the analysis also revealed downregulated pathways in the ligature groups. The first downregulated pathways are related to immune-associated signaling. For example, tumor necrosis factor–α signaling via NFκB was primarily reduced, a pathway inducing inflammation and cell survival (Hayden and Ghosh 2014). Moreover, interferon (IFN)–γ and IFN-α signaling reported to control mucosal epithelial homeostasis was also downregulated (Nava et al. 2010; Nassar et al. 2017) (Fig. 1E). A reduction was found in inflammatory and complement pathways and P53 activity, the latter being known to be suppressed during chronic inflammation (Gudkov et al. 2011). The analysis reveals decreased IL-2/STAT5 signaling, which promotes Treg cell differentiation (Laurence et al. 2007), while the reduction of the IL-6-JAK-STAT3 pathway suggests limited Th17 differentiation (Zhou et al. 2007). Comparing the Removal and Naïve groups (Fig. 1F) displayed an overall similar pattern of cellular pathways to those found between the Ligature and Naïve groups. However, the downregulation of the immune pathways was moderate while the tissue repair pathways were strengthened. Analysis of the Removal versus Ligature groups (Fig. 1G) identified only upregulated pathways that mirror the reduced pathways in the Ligature versus Naive groups. Moreover, the upregulation of pathways associated with tissue repair was observed, such as the epithelial-to-mesenchymal transition pathway (Zeisberg and Neilson 2009), mTORC1 signaling (Squarize et al. 2010), and IFNγ signaling (Kanno et al. 2019). These data demonstrate that LIP triggers rapid bone loss, which is subsequently halted due to the development of immunosuppression, while tissue repair processes are upregulated.
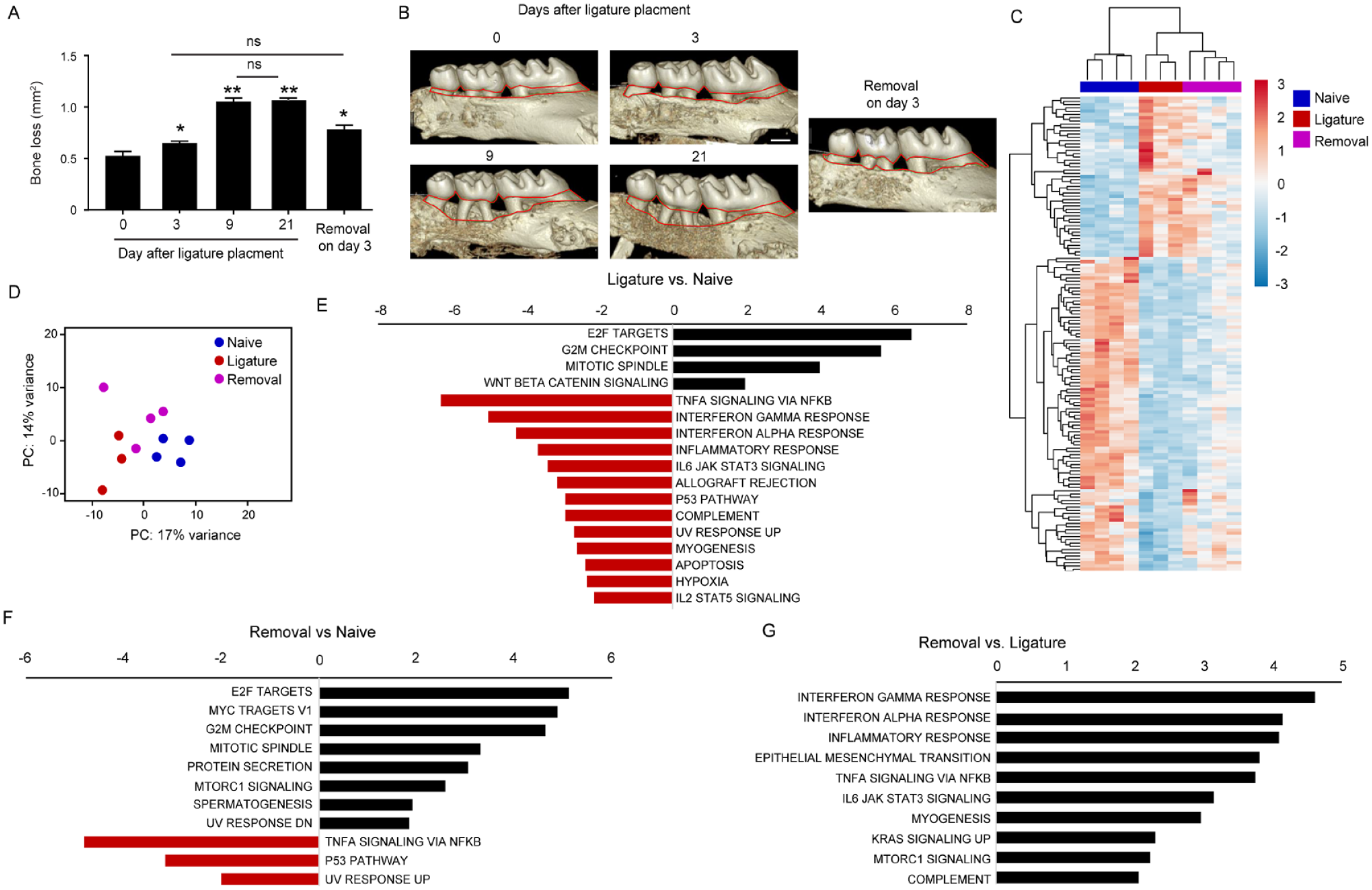
Ligature-induced periodontitis (LIP) induces rapid alveolar bone loss followed by gingival immunosuppressive conditions. LIP was induced in male B6 mice, and in part of the mice, the ligature was removed on day 3. (
LCs Mediate Innate Immunity during LIP and Inhibit Bone Loss
The RNAseq analysis demonstrates that the gingival epithelium responds to the ligature; thus, we asked whether LCs are involved in LIP-induced immunity. For this, LIP was induced in langerin–diphtheria toxin receptor (DTR) mice (Bennett et al. 2005), enabling the inducible ablation of gingival LCs using diphtheria toxin (DT). LCs were depleted 48 h before placing the ligature, and gingival innate immune responses were examined 3 d later. Flow cytometric analysis indicated that the frequencies of CD11b+ myeloid leukocytes increased in the ligatured gingiva, whereas the DT treatment abolished it (Fig. 2A–B and Appendix Fig. S1A). Within this population, the levels of neutrophils and Ly6Chigh monocytes decreased due to LC depletion. Since oral LCs regulate innate immune responses (Sparber et al. 2018), we quantified the expression of proinflammatory cytokines in the gingiva 24 h after LIP. As depicted in Figure 2C, LC depletion reduced the mRNA levels of Il23 and Il17a but not Il6 or Il1b. Despite this, 5 d after LIP, the frequencies of neutrophils and monocytes were similarly elevated in both LC-depleted and nondepleted mice (Appendix Fig. S1B). To examine the impact of LC depletion on bone loss, we used early and late depletion protocols, and the bone loss was quantified after 9 or 21 d of LIP, respectively (Fig. 2D). As demonstrated in Figure 2E, depletion of LCs resulted in elevated bone loss after both 9 and 21 d of LIP. In addition, the bone loss measured on day 21 of LIP was significantly higher than on day 9 when LCs were depleted. These findings suggest that LCs play a role in the induction of innate immune responses and the inhibition of bone loss during LIP.
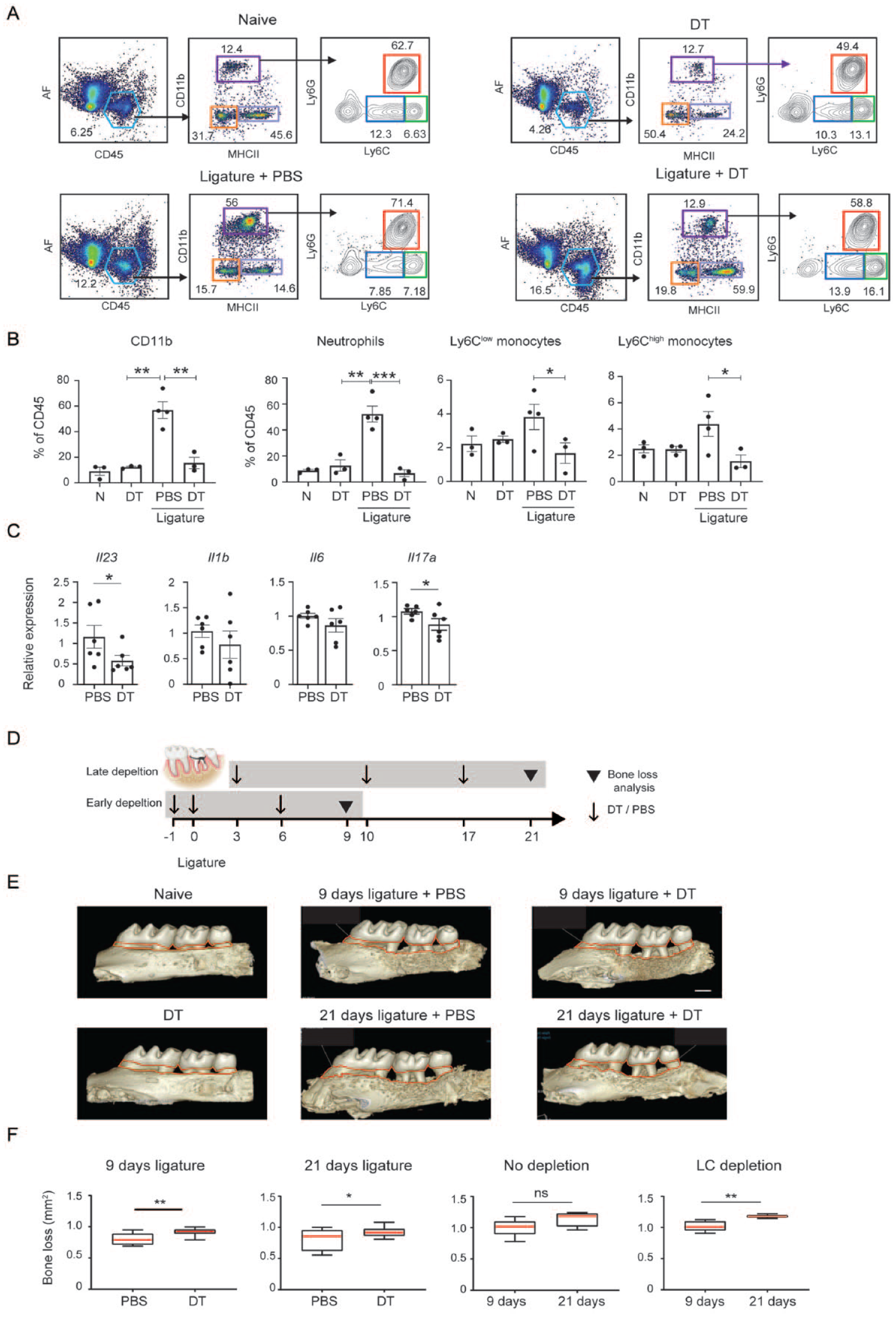
Langerhans cells (LCs) mediate innate immunity during ligature-induced periodontitis (LIP) and inhibit bone loss. Langerin–diphtheria toxin receptor (DTR) mice were treated with diphtheria toxin (DT) or phosphate-buffered saline (PBS) 48 h before initiating LIP; 3 d later, the gingival tissues were analyzed using flow cytometry. As a control, intact langerin-DTR mice were similarly treated with DT or PBS only. (
LCs and EpCAM+ DCs Rapidly Migrate to the Lymph Nodes during LIP and Monocyte-Derived EpCAM+ DCs Repopulate the Ligatured Gingiva
We next examined if the ligature promotes the migration of LCs to the lymph nodes (LNs) and whether the LCs repopulate the ligatured gingiva. To track LC migration, the gingiva was painted with fluorescein isothiocyanate (FITC) solution shortly before placing the ligature, and 3 d later, the LNs were analyzed (Fig. 3A). Gating on CD11c+MHCIIhi tissue-derived DCs and next on langerin+EpCAM+ cells (i.e., LCs), FITC-labeled LCs representing migratory LCs were detected in the LNs. The frequencies of migratory LCs were higher in the ligatured mice than in mice treated with FITC only, indicating that the ligature facilitates LC migration. EpCAM+ DCs, which are also located in the epithelium and give rise to LCs (Capucha et al. 2015; Capucha et al. 2018), also migrated to the LNs (Fig. 3A). Next, the repopulation of LCs in the gingiva was analyzed. As depicted in Figure 3B, while MHCII+CD11c+ DCs populated the gingiva, these cells did not fully differentiate into LCs, and instead, mainly EpCAM+ DCs developed. Accordingly, the mRNA levels of langerin (Cd207) were reduced in the gingiva during LIP (Fig. 3C). Using immunofluorescence analysis, cells stained positively for MHCII and langerin (i.e., LCs) were virtually absent in the gingiva at 9 d of LIP but were visualized when the ligature was removed earlier on the third day of LIP (Fig. 3D). Using Ms4a3TdT mice, enabling fate-mapping of monocyte-derived cells, the EpCAM+ DC population was found to originate from monocytic precursors (Fig. 3E). The inability of the monocytes to fully differentiate into LCs was not due to reduced expression of TGF-β and BMP7, cytokines driving steady-state LC differentiation (Capucha et al. 2018), as both cytokines were expressed in the ligatured gingiva (Fig. 3F, G). Taken together, early during LIP, the LCs migrate to the LNs while the ligature directs the differentiation of incoming monocytes to EpCAM+ DCs rather than to fully developed LCs.
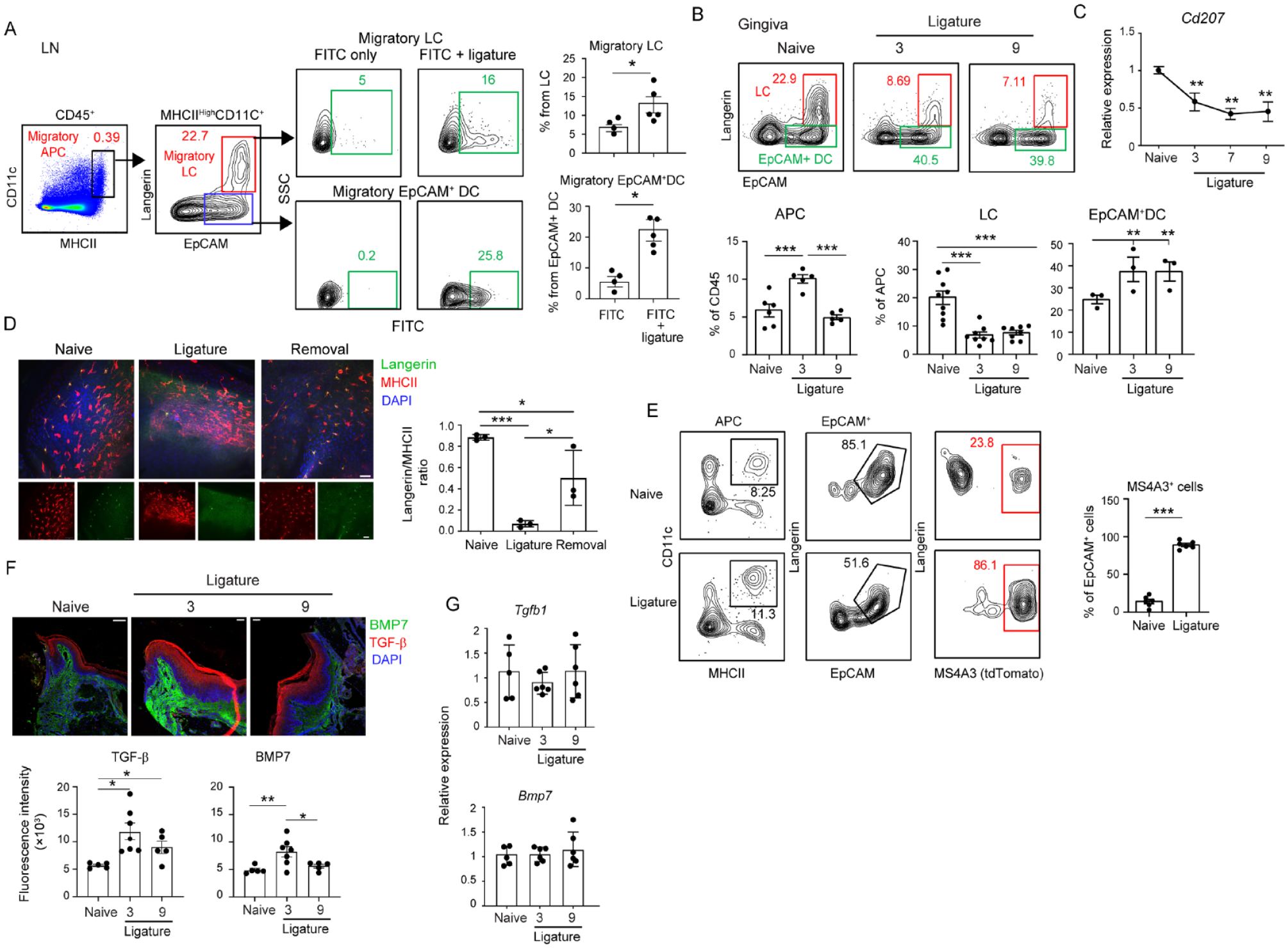
Migratory and repopulation capabilities of gingival epithelial antigen-presenting cells (APCs). The gingiva of B6 mice was painted with fluorescein isothiocyanate (FITC) solution before placing the ligature. (
LCs Mediate the Increase of Treg Cells during LIP
Treg cells were reported to prevent bone loss during murine LIP (Garlet et al. 2010; Greene et al. 2022). Since LCs are capable of polarizing T cells into Treg cells (Kautz-Neu et al. 2011; Arizon et al. 2012), we examined whether LCs affect Treg cells during LIP. First, we measured the frequencies of FOXP3+ CD4+ Treg cells in the ligatured gingiva. Higher percentages of Treg cells were found on day 7 of LIP compared with the naïve group, while a moderate reduction in the Treg population was observed on the ninth day (Fig. 4A). Quantification of the Foxp3 mRNA levels in the gingiva by quantitative polymerase chain reaction further substantiated these findings (Fig. 4B). LCs were next depleted by administering DT into langerin-DTR mice every 2 d during LIP, resulting in a reduction in both the total CD4+ T cells and the FOXP3+ Treg subpopulation (Fig. 4C). Since steady-state LCs largely disappeared from the gingiva on the third day of LIP and were replaced by EpCAM+ DCs, we next asked during which period of LIP the LCs influence Treg cells. To address this question, we initially measured the migration potential of LCs and EpCAM+ DCs on the third day of LIP. For this, we painted the ligatured gingiva with FITC on day 3 of LIP and analyzed the migrating DCs in the LNs 3 d later (Fig. 4D). For comparison, another group was FITC painted on day 0 of LIP, and the LNs were analyzed 3 d later. We first observed that the overall percentages of tissue-derived DCs (CD11c+MHCIIhi) were higher in the LNs on day 3 than on day 7 of LIP (Fig. 4D). Unlike tissue-derived DCs, no significant differences in the percentages of tissue-derived LCs (Langerin+EpCAM+) were found between the 2 groups. Nevertheless, analysis of FITC-labeled migratory cells further revealed that only the frequencies of FITC+ LCs, but not FITC+ EpCAM+ DCs, were reduced in the group painted on the third day of LIP compared with day 0. Next, LCs were depleted from langerin-DTR mice, either on day 0 or day 3 of LIP, and the frequencies of Treg cells were measured on day 7 (Fig. 4E). Flow cytometry analysis revealed a reduction in the gingival CD4+ T cells only when LCs were depleted on day 0 but not day 3 of LIP. Moreover, although the Treg cells were reduced in both depletion schedules, the impact of the depletion was more significant when LCs were depleted at the beginning of LIP than on day 7. Next, to determine whether Treg cells are induced in the LNs or expand locally during LIP, mice were administered FTY720, a sphingosine-1-phosphate receptor modulator that sequesters lymphocytes in LNs. The frequencies of Treg among CD4+ T cells were not changed, and Treg cells were positive for Ki67, a protein indicating cell proliferation (Appendix Fig. S4). However, the gingival CD4+ T cells were reduced by FTY720 treatment, resulting in an overall reduction of Treg cells in the ligatured gingiva. This suggests that Treg cells are induced in the LNs during LIP while LCs influence this process.
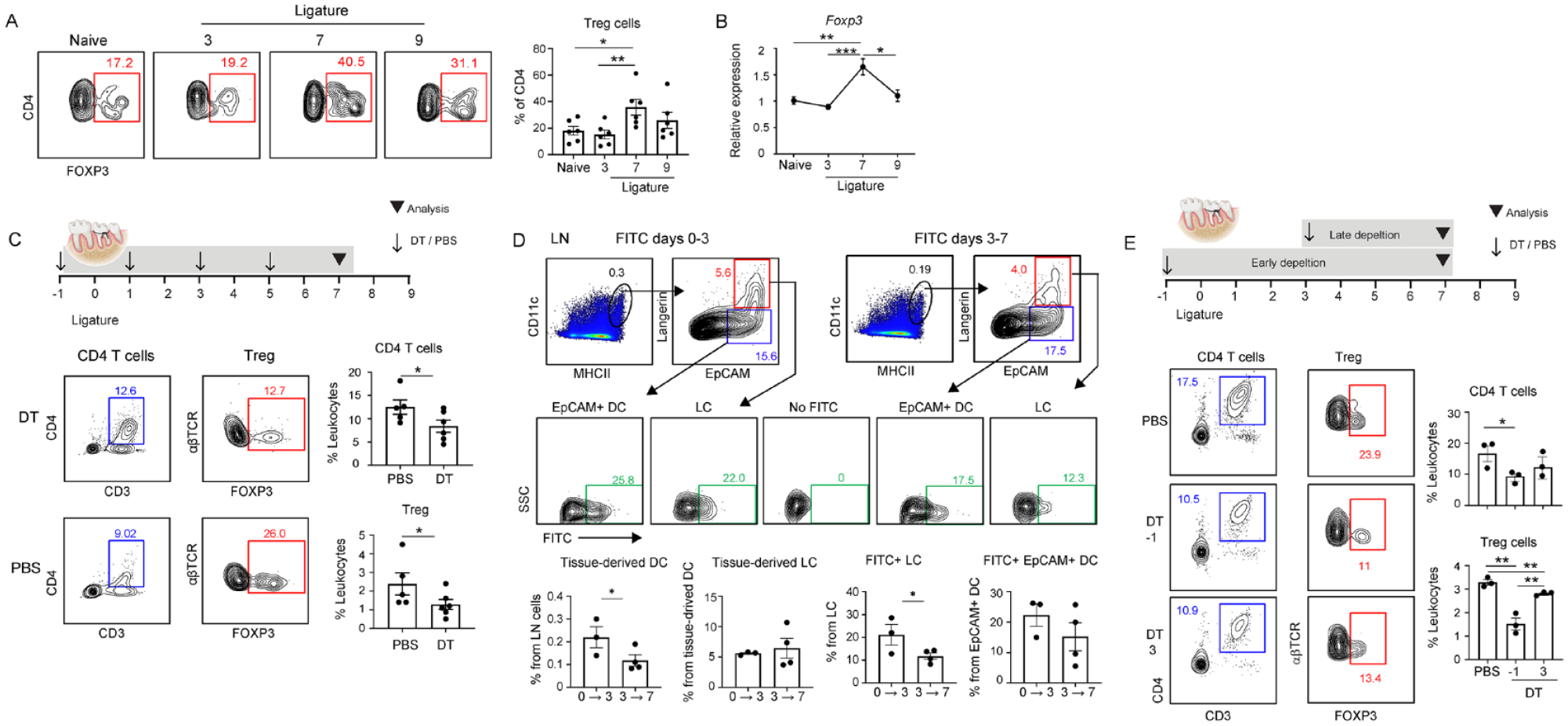
Langerhans cells (LCs) mediate the induction of Treg cells during ligature-induced periodontitis (LIP). (
RANKL Is Expressed by Gingival Epithelial Cells during LIP and Increases Treg Cell Levels
Since skin keratinocytes overexpress RANKL under inflammatory conditions and induce immunosuppression by expanding Treg cells (Loser et al. 2006), we asked whether RANKL is also expressed by gingival epithelial cells. The analysis revealed that during LIP, gingival epithelial cells upregulate RANKL expression, which was prevented by early removal of the ligature (Fig. 5A, B). RANKL also enhances LC development, as its presence in LC differentiation cultures increases the frequencies of LCs and EpCAM+ DCs (Appendix Fig. S2A). Next, we examined whether the epithelial LCs and EpCAM+ DCs express RANK. Essentially, all steady-state LCs express RANK (Fig. 5C). During LIP, the remaining LCs express RANK and the majority of EpCAM+ DCs, whereas most of the EpCAMneg DCs did not express this receptor (Fig. 5C). To examine if RANKL-RANK signaling affects the Treg cells, we depleted RANKL in vivo using monoclonal antibody either on day 1 and 3 or day 5 and 7 during LIP (Fig. 5D). Analysis of the gingiva on day 9 of LIP revealed that Treg cells were reduced in both depletion schedules (Fig. 5D). In contrast, the depletion of RANKL had no impact on the cervical LNs (Appendix Fig. S3). Depletion of RANKL also prevented the downregulated expression of Tnfa, Ifng, and Ifna, which was associated with gingival immunosuppression (Fig. 5E). No effect was found on Il17a expression, which was used as a control gene. To directly assess the impact of RANKL expression by gingival epithelial cells, we used transgenic mice overexpressing RANKL under the control of the keratin 14 promoter (K14-RANKL) (Loser et al. 2006). As illustrated in Figure 5F, the gingiva of K14-RANKL mice exhibited a 10-fold increase in Rankl expression compared with wild-type (WT) mice. Following the induction of LIP, K14-RANKL mice displayed less alveolar bone loss than the WT controls did (Fig. 5G). Flow cytometry analysis further revealed higher frequencies of Treg cells in the gingiva of ligatured K14-RANKL mice (Fig. 5H). Next, we investigated the impact of epithelial RANKL on the steady-state LC population of gingival LCs, revealing increased frequencies in the K14-RANKL mice (Appendix Fig. S5A). The mean fluorescence intensity of langerin on the LCs was elevated in these mice (Appendix Fig. S5B). Notably, the analysis also revealed increased frequencies of Treg cells under steady-state conditions (Fig. 4C). These findings suggest that upregulation of RANKL in the gingival epithelium increases Treg cell levels and mitigates LIP-associated bone loss.
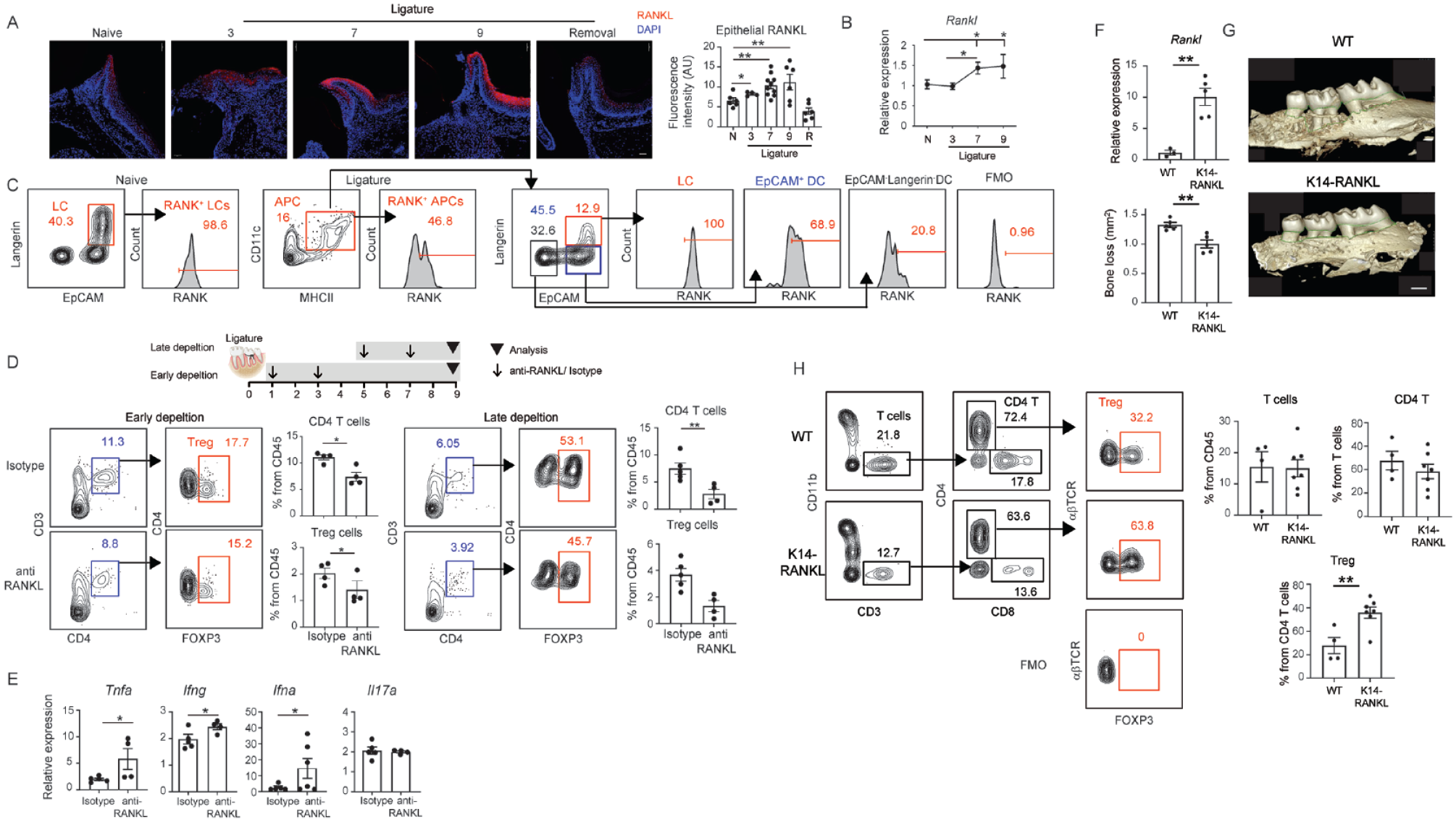
RANKL is expressed by gingival epithelial cells during ligature-induced periodontitis (LIP) and increases Treg cell levels. (
Discussion
This study proposes that in addition to its known pathological role in periodontitis, RANKL also has a protective immunological function. This could explain the pathological effect of anti-RANKL (denosumab) treatment in patients receiving it for the prevention of osteoporosis or bone metastasis (Ruggiero et al. 2014). These patients are at risk of developing osteonecrosis of the jaw, especially after tooth extraction, because the treatment might abolish the protective effect of RANKL. In agreement with this hypothesis, a recent study using the LIP model reported that reducing preexisting periodontal inflammation (i.e., by removing the ligature) before tooth extraction ameliorated medication-related osteonecrosis of the jawlike lesion in mice (Kim et al. 2018). Of note, RANKL inhibitors also induce osteonecrosis of the jaw in mice with periapical disease (Aghaloo et al. 2014), an oral disease in which RANKL expression is upregulated and required for Treg induction (Francisconi et al. 2018).
The ligature poses both mechanical and microbial challenges to the epithelium, prompting gingival LCs to respond to these epithelial insults. Gingival LCs mediate adaptive immunity to repair epithelial damage caused by early-life masticatory mechanical forces (Jaber et al. 2023). LCs also induce protective immunity upon infection with a periodontal pathogen (Arizon et al. 2012), which is capable of causing microbial dysbiosis (Lamont and Hajishengallis 2015). In the present study, LCs control LIP-induced innate immune responses, consistent with previous work (Sparber et al. 2018). While this suggests that LCs contribute to the bone loss associated with LIP, depletion of LCs inhibits this pathological process. This can be explained by the view that, as epithelial sentinels, LC-mediated innate immunity is likely beneficial for rapid epithelial repair and prevention of bacterial invasion. Nonetheless, the frequencies of neutrophils and monocytes eventually increased in the ligatured gingiva, suggesting the presence of a mechanism that accelerates neutrophil/monocyte recruitment and potentially exacerbates the pathology. As the local inflammation becomes chronic, the LCs mediate the increase of Treg cells to inhibit destructive inflammation. Treg cells can inhibit osteoclastogenesis by preventing the differentiation of late osteoclast precursors and regulating osteoclast function (Fischer et al. 2019). The impact of FTY720 treatment suggests that LCs induce Treg cells in the LNs, aligning with the view that the oral cavity does not support Treg generation at a steady state (Park et al. 2018). It is worth noting that changes in microbial stimuli occurring during LIP, as periodontal damage progresses and the epithelium detaches from the ligature, might also regulate LC/EpCAM+ DC function. This could contribute to alterations in local immunity, cessation of bone loss, and initiation of tissue repair mechanisms.
Expression of RANKL by gingival epithelial cells and its impact on local immunity signifies the importance of nonhematopoietic cells in orchestrating local immunity. Both LCs and EpCAM+ DCs are likely to interact with RANKL-expressing epithelial cells and induce Treg cells, as both cells express RANK and migrate to the LNs. This indicates that although EpCAM+ DCs are considered cells at a late stage of differentiation into LCs (Capucha et al. 2015; Capucha et al. 2018), they are also functional APCs. This is even more evident during inflammatory conditions when the gingival epithelium is repopulated by EpCAM+ DCs rather than fully developed LCs. It is hard to dissect the contribution of each subset to Treg induction since most LCs rapidly migrate to the LNs, a time in which RANKL is not strongly expressed by the epithelial cells. Nevertheless, steady-state LCs express RANK, and their depletion before ligature placement reduced the frequencies of gingival Treg cells. Moreover, steady-state LCs have a known capability to induce Treg cells upon activation (Kautz-Neu et al. 2011; Arizon et al. 2012). The EpCAM+ DCs, on the other hand, are the main APCs in the inflamed gingiva, and blocking RANKL signaling during this period results in a strong reduction of the Treg cells compared with blocking during the beginning of LIP. This suggests that EpCAM+ DCs are also important for the induction of Treg cells. A previous observation that RANKL blocking does not reduce preexisting Treg cells further substantiates this notion (Francisconi et al. 2018).
In conclusion, this work elucidates an underappreciated protective immunoregulatory activity for RANKL during experimental periodontitis, highlighting the complex role of this mediator in the disease. Hence, the ability of RANKL to modulate oral inflammation warrants further investigation as it may have clinical implications.
Author Contributions
Y. Netanely, O. Barel, R. Naamneh, Y. Jaber, Y. Saba, K. Zubeidat, O. Saar, L. Eli-Berchoer, contributed to conception, design, data acquisition, analysis, and interpretation, critically revised the manuscript; S. Yacoub, contributed to conception, design, data analysis and interpretation, critically revised the manuscript; S. Yona, A. Brand, T. Capucha, A. Wilensky, K. Loser, contributed to conception and design, critically revised the manuscript; B.E. Clausen, A.-H. Hovav, contributed to conception, design, drafted and critically revised the manuscript. All authors gave their final approval and agree to be accountable for all aspects of work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345241274370 – Supplemental material for Epithelial RANKL Limits Experimental Periodontitis via Langerhans Cells
Supplemental material, sj-docx-1-jdr-10.1177_00220345241274370 for Epithelial RANKL Limits Experimental Periodontitis via Langerhans Cells by Y. Netanely, O. Barel, R. Naamneh, Y. Jaber, S. Yacoub, Y. Saba, K. Zubeidat, O. Saar, L. Eli-Berchoer, S. Yona, A. Brand, T. Capucha, A. Wilensky, K. Loser, B.E. Clausen and A.-H. Hovav in Journal of Dental Research
Footnotes
Acknowledgements
We thank Yuval Nevo, Inbar Plaschkes, and Hadar Benyamini for the bioinformatic analysis.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Israel Science Foundation grant 2272/20 (A.-H.H) and the German Science Foundation (DFG, Deutsche Forschungsgemeinschaft) grant CL419/2-2 (B.E.C.).
A supplemental appendix to this article is available online.
Data Availability
The RNA-seq data generated in this study will be deposited in the NCBI Gene Expression Omnibus GEO database. All data are available in the main text and the supplementary materials.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.