
Abstract
Histone methylation assumes a crucial role in the intricate process of enamel development. Our study has illuminated the substantial prevalence of H3K4me3 distribution, spanning from the cap stage to the late bell stage of dental germs. In order to delve into the role of H3K4me3 modification in amelogenesis and unravel the underlying mechanisms, we performed a conditional knockout of Ash2l, a core subunit essential for the establishment of H3K4me3 within the dental epithelium of mice. The absence of Ash2l resulted in reduced H3K4me3 modification, subsequently leading to abnormal morphology of dental germ at the late bell stage. Notably, knockout of Ash2l resulted in a loss of polarity in ameloblasts and odontoblasts. The proliferation and apoptosis of the inner enamel epithelium cells underwent dysregulation. Moreover, there was a notable reduction in the expression of matrix-related genes, Amelx and Dspp, accompanied with impaired enamel and dentin formation. Cut&Tag-seq (cleavage under targets and tagmentation sequencing) analysis substantiated a reduction of H3K4me3 modification on Shh, Trp63, Sp6, and others in the dental epithelium of Ash2l knockout mice. Validation through real-time polymerase chain reaction, immunohistochemistry, and immunofluorescence consistently affirmed the observed downregulation of Shh and Sp6 in the dental epithelium following Ash2l knockout. Intriguingly, the expression of Trp63 isomers, DNp63 and TAp63, was perturbed in Ash2l defect dental epithelium. Furthermore, the downstream target of TAp63, P21, exhibited aberrant expression within the cervical loop of mandibular first molars and incisors. Collectively, our findings suggest that ASH2L orchestrates the regulation of crucial amelogenesis-associated genes, such as Shh, Trp63, and others, by modulating H3K4me3 modification. Loss of ASH2L and H3K4me3 can lead to aberrant differentiation, proliferation, and apoptosis of the dental epithelium by affecting the expression of Shh, Trp63, and others genes, thereby contributing to the defects of amelogenesis.
Introduction
Developmental defects of enamel (DDEs) are prevalent clinical hard tissue disease. The incidence of DDEs for primary dentition varies from 15% to 49% globally (Xu et al. 2022), whereas for permanent dentition, it can reach as high as 21% to 99.6% (Vargas-Ferreira et al. 2018). These defects lead to a series of problems, including tooth wear, caries, pulpitis, and tooth loss, impairing masticatory and aesthetic functions (Costa et al. 2017). Gaining a deeper understanding of the pathogenesis of DDEs offers the promise of formulating an early intervention program. Hence, it becomes imperative to comprehend the underlying mechanisms of amelogenesis.
Amelogenesis is a highly intricate and well-organized process that involves proliferation and differentiation of the thickened dental epithelium to form the enamel organ, which undergoes several stages: bud, cap, and bell stage. Typical bell stage enamel organ is composed of 4 layers of cells, including the inner enamel epithelium (IEE), outer enamel epithelium (OEE), stellate reticulum (SR), and stratum intermedium (SI). During the late bell stage, the IEE differentiates into ameloblasts that synthesize and secrete extracellular matrix, such as AMELX (amelogenin X-linked). Formation of enamel and dentin is mainly regulated by the crosstalk between the dental epithelium and dental mesenchyme. Several classical signaling pathways, such as bone morphogenetic protein (BMP), fibroblast growth factor (FGF), hedgehog (Hh), and Wnt, are involved in these processes (Balic and Thesleff 2015). These pathways modulate the expression of key downstream molecules, finely tuning the development of the dental epithelium and enamel formation by influencing proliferation, differentiation, and stemness of dental epithelial stem cells (Landin et al. 2012).
Emerging studies have highlighted the crucial role of epigenetics in tooth development (Townsend et al. 2005; Suda et al. 2010). For instance, our previous research demonstrated that disturbance of N6-methyladenosine (m6A) RNA methylation in the dental epithelium can result in DDEs in mice (Xie et al. 2022). As one of the most common epigenetic modifications, H3K4 methylation is mainly catalyzed by the lysine-specific methyltransferases subclass 2 (KMT2) complex, including KMT2A-D (also known as MLL1–4; mixed-lineage leukemia 1–4) and KMT2F-G (also known as SET1A-B) (Chen et al. 2012; Hyun et al. 2017; Bochyńska et al. 2018). Disruption of H3K4 methylation might relate to DDEs in humans. Mutation in KMT2D has been associated with Kabuki syndrome, a multisystemic disorder characterized by various developmental defects, including a series of dental anomalies, such as enamel defects (Cudzilo and Czochrowska 2018). It is also reported that KMT2D deficiency disturbs proliferation and differentiation of the ameloblast-like cell line LS8, partially via the Wnt signaling pathway (Pang et al. 2021). However, the specific mechanism by which H3K4 methylation dysfunction contributes to DDEs in vivo is still not fully understood and requires further investigation.
To function properly, the KMT2 complex requires a suitable platform provided by the WRAD complex, which is composed of 4 core subunits: WDR5 (WD repeat domain 5), RbBP5 (retinoblastoma binding protein 5), ASH2L (absent, small, or homeotic 2-like), and DPY30 (dumpy-30) (Hyun et al. 2017). Since KMT2 contains 6 different enzymes with functional redundancy, it may not be the best choice to study the specific role of H3K4 methylation. Among the 4 core subunits of WRAD in the KMT2 complex, ASH2L is essential for H3K4 methylation (Barsoum et al. 2022). Conditional knockout of Ash2l in mouse neural progenitor cells leads to reduced H3K4me3, resulting in structural abnormalities of mouse neural cortex and decreased nerve number by the Wnt/β-catenin signaling pathway (Li et al. 2019). Similarly, Ash2l knockout in the hematopoietic system leads to downregulation of mitosis-related gene expression, causing loss of hematopoietic stem cells in mice and death after birth (Lüscher-Firzlaff et al. 2019). However, whether ASH2L and H3K4me3 are involved in enamel development remains unknown.
To address the aforementioned issues, conditional Ash2l knockout mice in the dental epithelium were generated. Our study aims were to investigate whether elimination of H3K4me3 leads to severe amelogenesis imperfecta by conditional knockout of Ash2l in the dental epithelium and to identify the possible downstream regulated genes. Our findings will uncover the important role of ASH2L and its mediated H3K4me3 in amelogenesis and provide novel insights into the mechanisms underlying enamel development.
Methods
Experimental details are available in the Appendix material. Raw data of RNA-seq and Cut&Tag-seq (cleavage under targets and tagmentation sequencing) were uploaded to BioProject PRJNA975737. Ash2lflox/flox mice were derived from Dr. Xiaozhong Peng (School of Basic Medicine, Peking Union Medical College, Beijing, China) (Li et al. 2019). The animal studies conform the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Results
Conditional Knockout of ASH2L in Mice Exhibited Developmental Defects of Dental Germ at the Late Bell Stage
ASH2L was found highly expressed in mice enamel organ at E14.5 (embryonic day 14.5, cap stage), E16.5 (embryonic day 16.5, early bell stage), E18.5 (embryonic day 18.5, late bell stage), and P0 (postnatal day 0, late bell stage) (Appendix Fig. 1A–E). At the same time, the distribution of H3K4me3 was also detected in E14.5, E16.5, E18.5, and P0 enamel organ (Appendix Fig. 1F–J), which is consistent with previous research findings (Zheng et al. 2014). These results indicated that ASH2L and H3K4me3 might be involved in enamel development.
To investigate the cell-autonomous function of H3K4me3, we generated dental epithelium–specific mutants of Ash2l by intercrossing Ash2lflox/flox mice with K14-Cre mice (Fig. 1A, Appendix Fig. 3A, B). The Ash2lflox/flox mice were crossed with K14-cre transgenic mice to generate Ash2lflox/flox, K14-Cre mice (named cko). Littermates of the cko mice, identified as heterozygous, served as control subjects (named Ctrl). The cko mice did not have a notably decreased body weight and obvious oral-maxillofacial deformities at P0 (Fig. 1D, E). However, they experienced postnatal mortality within 24 h. Upon analyzing lung tissue sections, we observed a pronounced thickening of the alveolar epithelium in the cko mice (Appendix Fig. 2), suggesting that this condition could have contributed to their postnatal demise.
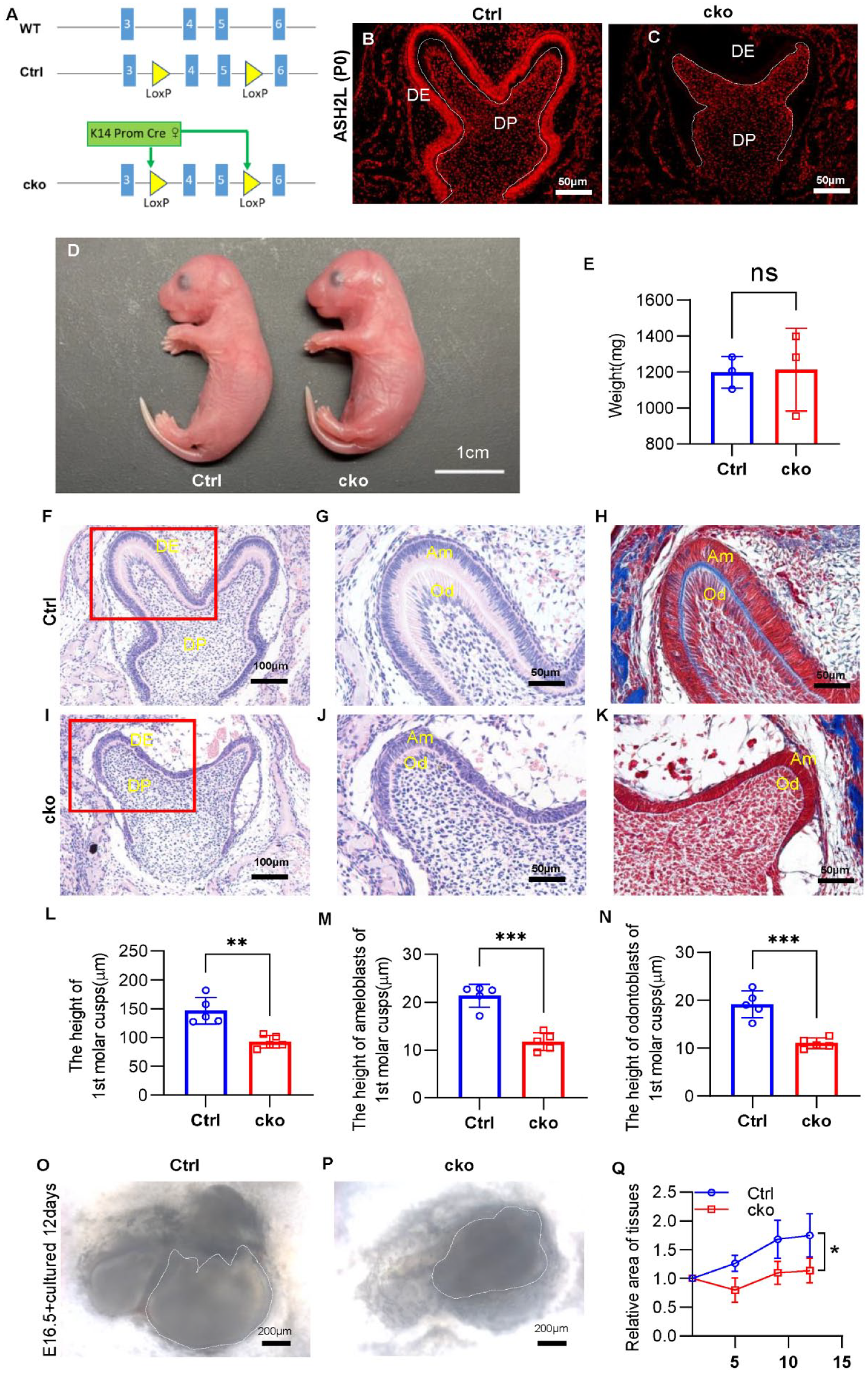
Deletion of Ash2l in the dental epithelium impairs dental germ development. (
The result of immunofluorescence proved that cko mice failed to express ASH2L in the dental epithelium. Nevertheless, the ASH2L protein in other tissues such as dental mesenchyme remained undisturbed (Fig. 1B, C, Appendix Fig. 3C–H). However, the size of the mandibular first molars and height of cusps were decreased in cko mice at P0 (Fig. 1F, I, L). At the late bell stage, IEE differentiates into ameloblasts and undergoes a transformation in cell morphology from cuboidal to tall and highly polarized columnar cells (Balic and Thesleff 2015). As we could see, the morphology of ameloblasts was highly polarized and tall columnar in Ctrl mice, while the height and polarization of ameloblasts were greatly reduced in cko mice. The SI, which was neatly arranged outside of the IEE in Ctrl dental germ, was disorganized in cko. In addition, the odontoblasts of cko mice were also less polarized than Ctrl (Fig. 1G, J, M, N, Appendix Fig. 4A, B). In line with the cell morphological change, greatly reduced and irregular matrix was seen between IEE and odontoblasts at molar cusps and incisors in cko mice compared with Ctrl mice (Fig. 1H, K, Appendix Fig. 4C–G).
To investigate which stage was affected, we sectioned mandibular first molars at the cap and early bell stages, respectively. It was found that abnormal changes happened in the late bell stage, and the early bell stage and cap stage were unaffected (Appendix Fig. 3I–P). To exclude the possible influence of internal environmental changes on dental germs, we cultured mandibular first molars of E16.5 in vitro. Compared with Ctrl tissue, the growth rate was lower, morphology of the cusp was not obvious, and the edge of the dental germ was indistinct in cko (Fig. 1O–Q). These suggest that inactivation of ASH2L leads to disorders of dental germ at the late bell stage.
ASH2L Deletion in the Dental Epithelium Disturbed the Differentiation, Proliferation, and Apoptosis of Dental Germs
AMELX and dentin sialophosphoprotein (DSPP) are extracellular matrix proteins; the former is reported expressed in secretory ameloblasts and odontoblasts, and the latter is expressed in presecretory ameloblasts and odontoblasts (Verdelis et al. 2016; Isono et al. 2021). Real-time polymerase chain reaction (PCR) and immunohistochemistry analyses unveiled a discernible decline in AMELX expression within the dental epithelium, and a reduction in DSPP levels was observed in the dental papilla (Fig. 2A–H). Besides, as a marker of odontoblast differentiation (Chen et al. 2022), the expression of P21 was decreased in cko odontoblasts (Fig. 4Q). In terms of morphology and function, it can be found that the differentiation of ameloblasts and odontoblasts was impaired because of the absence of ASH2L in the dental epithelium.
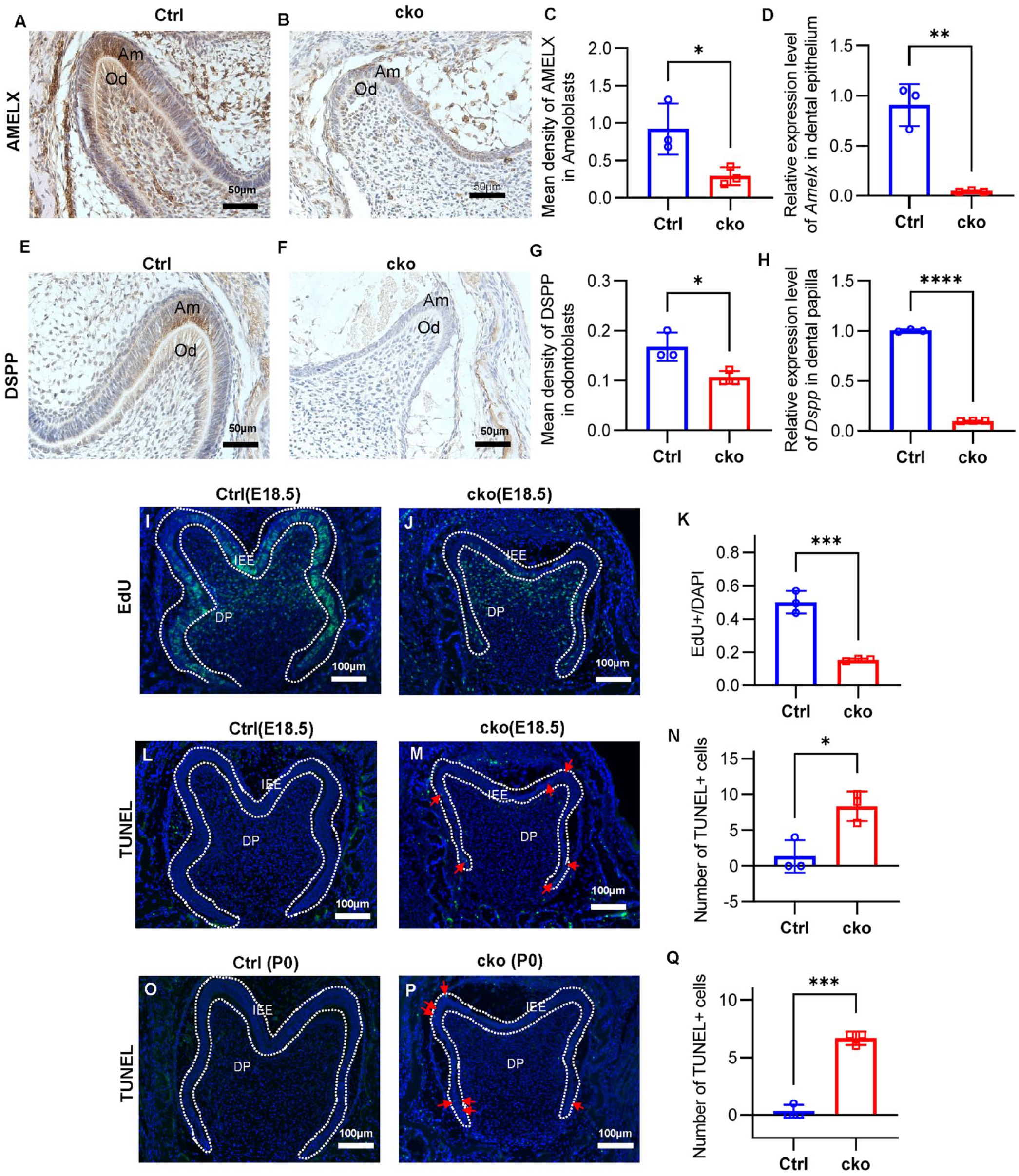
Diminishment of Ash2l leads to abnormal differentiation, proliferation, and apoptosis of dental germs. (
The dental epithelium continuously multiplies and folds during formation of enamel organs (Lacruz et al. 2017), so we detected the proliferation and apoptosis of the dental epithelium by EdU (5-ethynyl-2′-deoxyuridine) and TdT mediated dUTP nick end labeling (TUNEL) staining. The results showed that the number of EdU-positive cells was greatly reduced in the IEE of cko mice (Fig. 2I–K) at the late bell stage. Besides, there were very few apoptosis cells in the IEE of Ctrl mice at the late bell stage, while several TUNEL positive cells were observed in the IEE of cko mice (Fig. 2L–Q). The EdU and TUNEL staining proved that loss of ASH2L in the dental epithelium depressed proliferation and resulted in apoptosis of the IEE, which might have led to the decreased size of molars.
Loss of ASH2L Disturbed the Expression of Amelogenesis-Related Genes in the Dental Epithelium
RNA sequencing was conducted to identify the molecular changes in P0 mandibular molar germs resulting from Ash2l deletion. There were 919 differentially expressed genes, including 588 downregulated and 331 upregulated genes in cko mandibular first molars compared with Ctrl (Fig. 3A, Appendix Fig. 5A, B). Gene Ontology (GO) enrichment analysis for differentially expressed genes was mainly gathered in skin development, epidermis development, and others. As for downregulated genes, amelogenesis, enamel mineralization, and tooth mineralization were included in the top 10 biological processes (Fig. 3B, Appendix Fig. 5C). Significant downregulation of genes closely associated with ameloblast and odontoblast differentiation, including Amelx, Dspp, Dmp1, Sp6, Tbx1, Fzd3, Fzd6, Shh, and Trp63, was validated by real-time PCR in CKO mandibular first molars (Fig. 3C). Expression of Fzd3, Fzd6, Sp6, Tbx1, Shh, and Dmp1 was also proved decreased in P0 or E18.5 CKO dental epithelium and dental papilla, respectively (Fig. 3D, E, Appendix Fig. 5D–I).
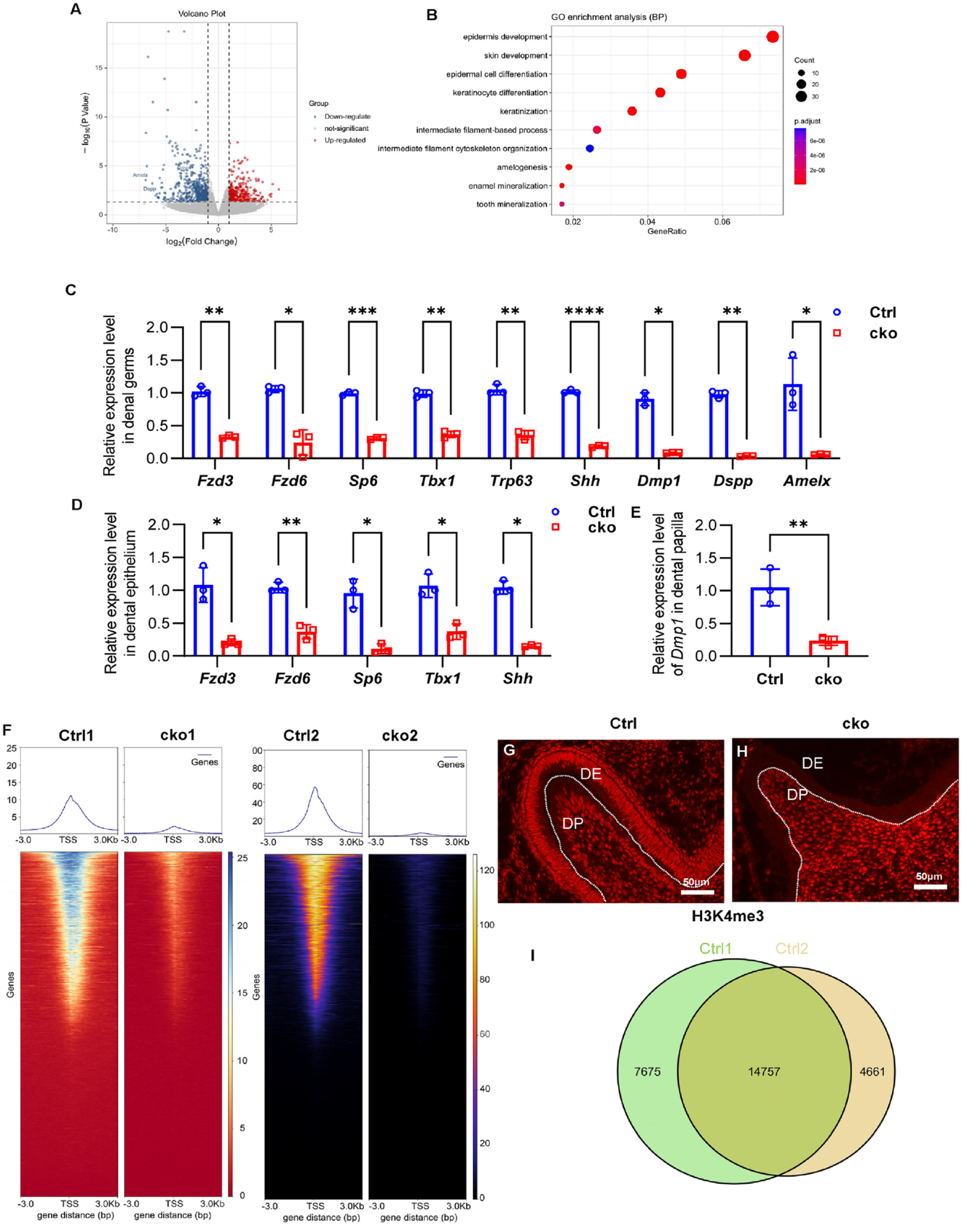
Ash2l inactivation alters gene expression and H3K4me3 establishment. (
ASH2L plays a crucial role in regulating the establishment of H3K4me3, which is typically located at the gene promoter region and involved in activating gene expression (Barsoum et al. 2022). The immunofluorescence revealed that deletion of Ash2l led to the disruption of H3K4me3 establishment in the dental epithelium (Fig. 3G, H, Appendix Fig. 6). To investigate the mechanism underlying the regulation of amelogenesis by ASH2L, we performed Cut&Tag-seq to reveal the H3K4me3 landscape of late bell stage enamel organs. The filtered read profiling revealed a significant reduction in reads around the transcription start site (TSS) (Fig. 3F). The sparse enrichment analysis for Cut&Tag peak caller identified 22,432 and 19,418 peaks in 2 Ctrl replicates, respectively, with a high overlap of 14,757 peaks (Fig. 3I). It was observed that many genes related to amelogenesis, such as Shh, Trp63, Sp6, Tbx1, Fzd3, Fzd6, Stim1, Wnts, Bmps, Fgfs, and Nothchs (Balic and Thesleff 2015), were modified with the H3K4me3 (Fig. 4A–C, G, Appendix Fig. 7, Appendix Fig. 8K), indicating the involvement of ASH2L and H3K4me3 modification in the regulation of amelogenesis.
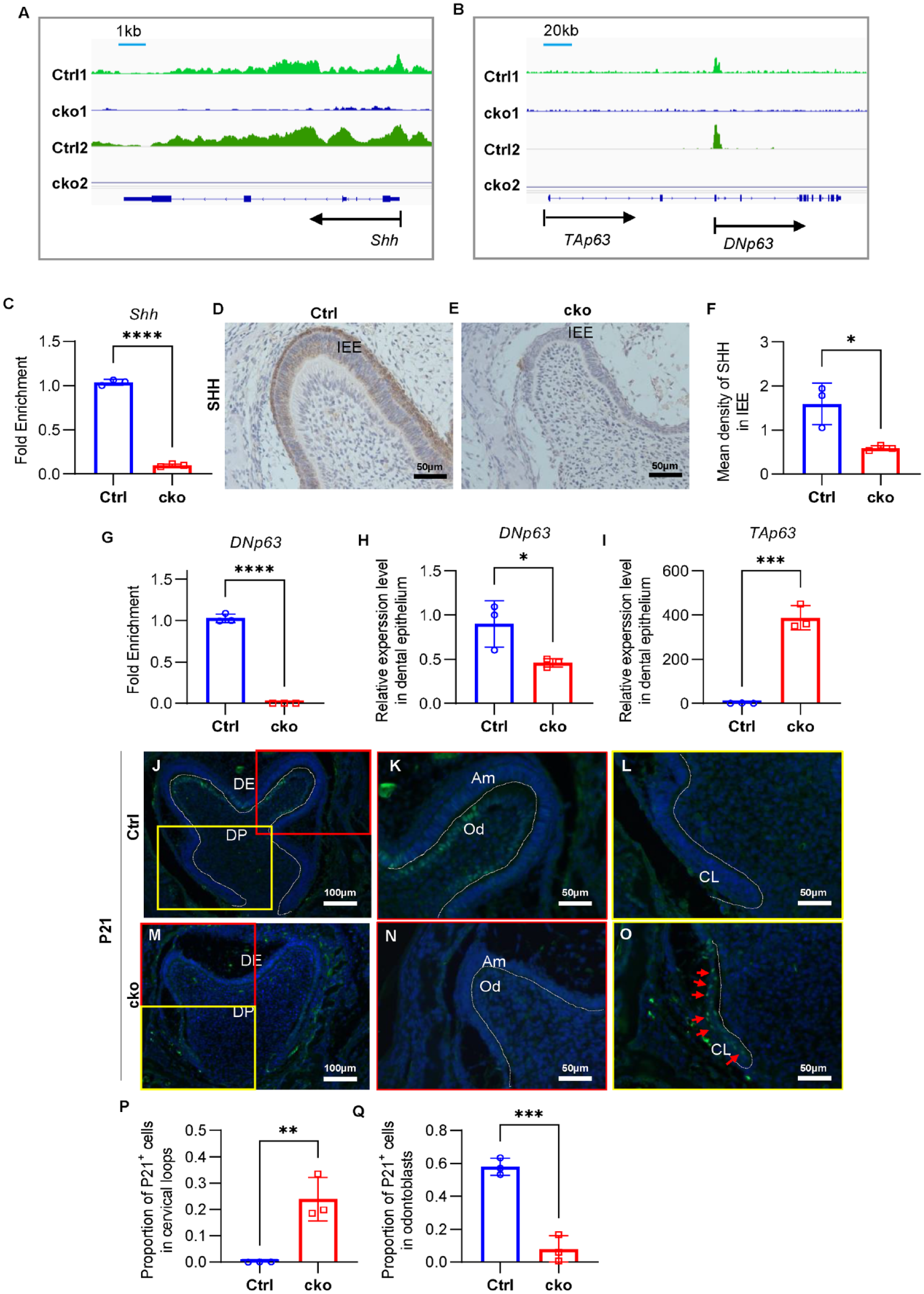
ASH2L regulates SHH and Trp63 expression by H3K4me3 modification. (
Notably, among these genes, a pronounced reduction in expression was observed for Sp6, Tbx1, Fzd3, Fzd6, Shh, and Trp63, implying the potential regulatory role of ASH2L in orchestrating the establishment of H3K4me3 on these specific genes, consequently influencing their expression. Besides, we also found that Amelx, Dspp, and Dmp1 were not modified by H3K4me3. This suggested the observed decrease in expression of these genes could potentially be attributed to functional alterations in ameloblasts and odontoblasts.
To further validate the role of ASH2L and H3K4me3 in amelogenesis, we assessed the expression of SHH and SP6 in IEE, critical for regulating proliferation and differentiation of ameloblasts (Gritli-Linde et al. 2002; Nakamura et al. 2008). SHH and SP6 were observed to be downregulated within the IEE in the late bell stage, which aligns consistently with the outcomes of real-time PCR analyses (Fig. 4D–F, Appendix Fig. 8A–J, L). Interestingly, downregulation of SHH was observed only in cko mandibular first molars at E18.5 and P0 by immunohistochemistry, coinciding with the abnormal phenotype at late bell stage.
In addition, we examined the expression of P63 (coded by Trp63), a key regulator of epithelial development (Laurikkala et al. 2006). Due to different TSS, Trp63 generates 2 transcripts, TAp63 and DNp63 (Appendix Fig. 9A), with DNp63 predominantly expressed in the dental epithelium (Little and Jochemsen 2002). H3K4me3 was found on the promoter region of DNp63 (Fig. 4B), which indicated H3K4me3 mainly activates the expression of DNp63. Our real-time PCR results showed that DNp63 was downregulated in the enamel organ of cko mandibular first molars compared to Ctrl at the late bell stage (Fig. 4H, Appendix Fig. 9B). Interestingly, we found TAp63 was expressed at low levels in the enamel organ of Ctrl mandibular first molars as reported, but its expression was significantly increased in the cko enamel organ at the late bell stage (Fig. 4I, Appendix Fig. 9C). P21, an important downstream target of TAp63 (Suzuki et al. 2015), was abnormally expressed in the cervical loop region of cko mandibular first molars and incisors compared to Ctrl mice (Fig. 4J–P, Appendix Fig. 9D–G).
Taken together, ASH2L modulates the expression of genes, including Shh, Sp6, Trp63, and others, by H3K4me3 modification.
Discussion
Histone methylations play a vital role in tooth development by modifying genes with activating or silencing marks, thereby finely tuning related gene expression. Bivalent histone modifications of H3K4me3 and H3K27me3 are known to be widely imprinted on the genome of pluripotent stem cells, regulating stemness and multidirectional differentiation potential of stem cells through precise temporal and spatial expression of genes (Li et al. 2018). Recent studies have also shown that H3K4me3 and H3K27me3 are distributed temporally and spatially in the enamel organ. Specifically, H3K4me3, as an active marker of gene transcription, is detected from the bud to bell stage enamel organ. In contrast, H3K27me3, a silencing marker of gene transcription, only exists in the bud and cap stage enamel organ (Zheng et al. 2014). Previous studies have been proved that H3K27me3 plays a significant role in amelogenesis (Li et al. 2021; Yu et al. 2022). Research, including our own findings, has demonstrated that H3K4me3 is consistently imprinted in mouse enamel organs. It has been proved that H3K4me3 plays a critical role in differentiation and cell cycle of ameloblasts in vitro (Pang et al. 2021). However, before this study, it remained largely unknown how H3K4me3 modulates the cell lineage and fate determination during amelogenesis in vivo.
H3K4 methylation is associated with open chromatin and is primarily involved in activating gene transcription. Specifically, H3K4me3 mainly marks the promoters, typically residing near the TSS, and is associated with promoter accessibility. ASH2L has been identified as a crucial subunit involved in the formation of H3K4me3 (Bochyńska et al. 2018). In our research, Ash2l was knocked out in the dental epithelium to explore the potentially essential role of H3K4me3 in amelogenesis. Our findings illuminated that loss of Ash2l in the dental epithelium impeded ameloblast formation, subsequently resulting in enamel dysplasia. This observation strongly suggests the involvement of H3K4me3 in the regulation of amelogenesis.
As an active marker on promoters, H3K4me3 is mainly related to the activation of gene expression. So, we hypothesize that disestablishment of H3K4me3 on the genome of the dental epithelium thus regulates amelogenesis. As we know, the proliferation and differentiation of the dental epithelium are mainly regulated by multiple signaling pathways, such as Shh, Wnt, Fgf, Bmp, and transcription factors, including P63, P21, TBX1, SP6, and others (Smith et al. 2017). Our results revealed prevalent H3K4me3 modification on the genome of the dental epithelium, and many genes related to amelogenesis are modified.
Among these genes, the expression of Fzd3, Fzd6, Tbx1, Sp6, Shh, and Trp63 significantly decreased. Fzd3 and Fzd6 are primarily associated with planar cell polarity (Obara et al. 2017), which coincides with the observed loss of polarization in ameloblasts and odontoblasts. Defects of Tbx1 lead to the absence of enamel but have less effect on the formation of dentin (Catón et al. 2009). We also found that the phenotype of cko mice exhibited in ameloblast and odontoblast development is most similar to the phenotypes observed in Shh defect mice (Dassule et al. 2000). As a key regulator of amelogenesis, SHH is restricted in the dental epithelium, and loss of SHH leads to unpolarized ameloblasts and enamel hypoplasia with disturbed odontoblasts and dentin (Gritli-Linde et al. 2002; Hosoya et al. 2020). SP6 is also an important regulator of amelogenesis. Studies have reported that it promotes the proliferation and differentiation of the dental epithelium (Nakamura et al. 2008). Specially, the proteins SHH and SP6 were found to be decreased in cko dental epithelium at the late bell stage.
P63 is a key regulator of ectodermal development, and its absence can arrest tooth development at the dental lamina stage (Laurikkala et al. 2006). P63 likely carries out its function by controlling the expression of genes crucial for stem cell properties (Yang et al. 2006). P21 is reported as one of the best-characterized downstream targets of P63. The functional equilibrium between TAp63 and DNp63 isoforms regulates P21 expression, which influences the proliferative potential of epidermal progenitor cells (Suzuki et al. 2015). DNp63 is related to high proliferative activity, while TAp63 activates the expression of P21, thus inhibiting cell proliferation (Little and Jochemsen 2002). Our results revealed H3K4me3 binding sites enriched on the TSS of DNp63, and accordingly, the expression of DNp63 decreased in the CKO dental epithelium. Interestingly, we observed an abnormally high expression of TAp63 in the dental epithelium, along with ectopic expression of P21 in the CKO cervical loop of dental germs. It is speculated that disrupted balance between TAp63 and DNp63 could potentially be linked to the diminished proliferation in the IEE via regulating P21.
Tooth development is regulated by reciprocal communication between the dental epithelium and underlying mesenchyme, involving signal molecules such as BMPs, Wnts, FGFs, and Hedgehogs (Balic and Thesleff 2015). Interruption of signaling interaction between the dental epithelium and mesenchyme could result in developmental defects of enamel or dentin (Imhof et al. 2020). For instance, defects of SHH and SP6 in the dental epithelium lead to odontoblast dysdifferentiation and abnormal dentin formation (Nakamura et al. 2017; Hosoya et al. 2020). In our research, the differentiation of odontoblasts and formation of dentin were also damaged, most likely due to the affected interaction between the dental epithelium and mesenchyme caused by disrupted signaling in the dental epithelium resulting from ASH2L deletion.
However, our study has some limitations that should be considered. Although we deleted ASH2L and its mediated H3K4me3 in the dental epithelium at the cap stage, it is hard to explain why the observed phenotype mainly appeared during the late bell stage. In addition, while there are many genes modified by H3K4me3, it remains unclear why the deletion of ASH2L does not affect the expression of most of these genes. Other histone modifications such as H3K27me3 are also involved in amelogenesis (Yu et al. 2022), and it remains unknown whether other modifications on the genome change simultaneously to maintain certain gene expression levels after H3K4me3 modifications are lost in the dental epithelium. These issues require further investigation in future studies.
Importantly, our study confirms, for the first time, the direct involvement of H3K4me3 in amelogenesis. The deficiency of ASH2L in the dental epithelium led to the perturbation of H3K4me3 on genes associated with amelogenesis, consequently causing abnormal expression of Shh, Trp63, and others. This disturbance ultimately led to reduced proliferation and elevated levels of apoptosis in the IEE, along with disrupted differentiation in ameloblasts and odontoblasts. As a consequence, abnormalities in the formation of enamel and dentin emerged as the final outcome (Fig. 5). In summary, our results highlight the function of ASH2L and its mediated H3K4me3 modification in amelogenesis.
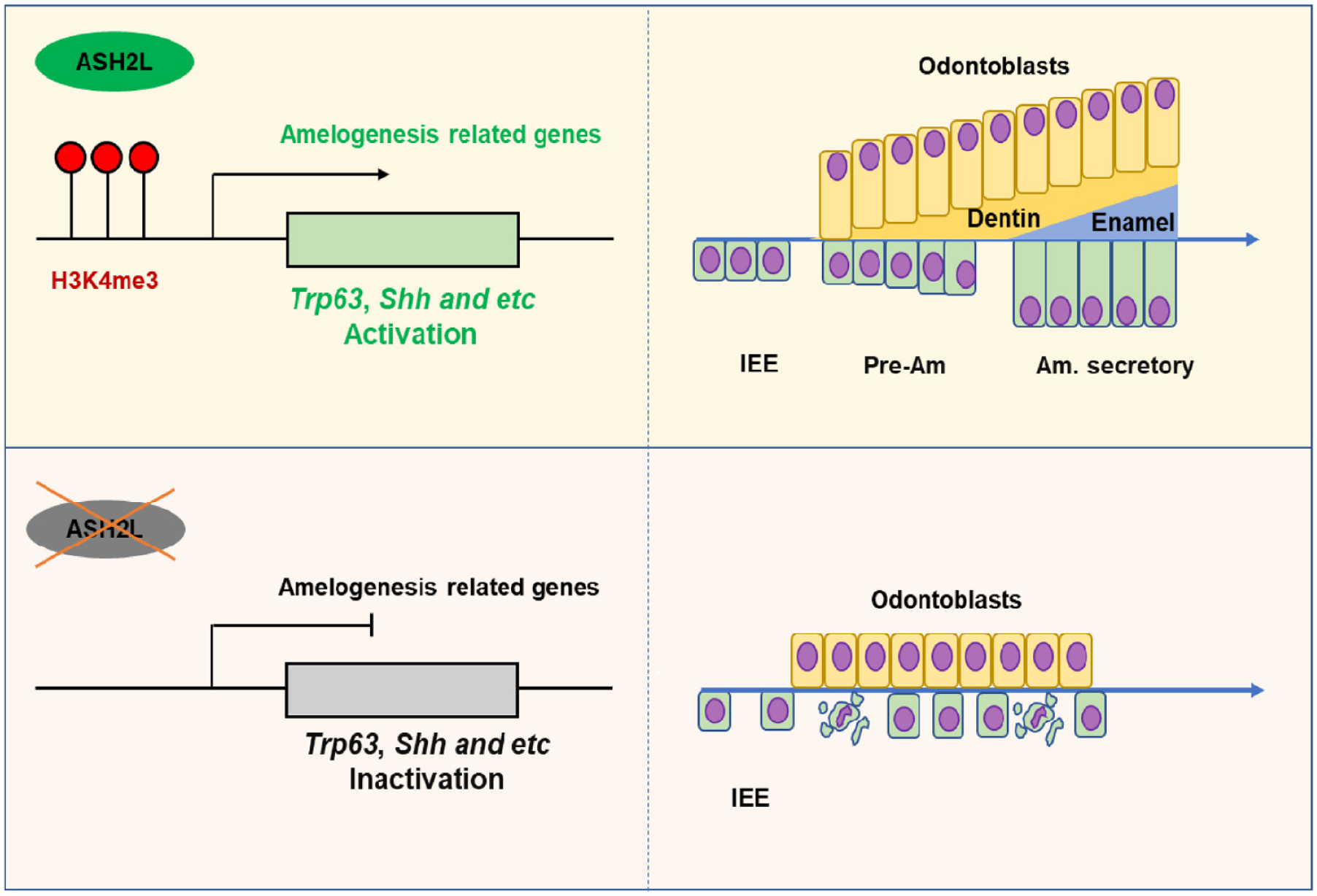
The role of ASH2L and H3K4me3 in amelogenesis regulation. Ash2l inactivation suppresses H3K4me3, disrupting the expression of Shh, Trp63, and other genes, leading to disturbances in amelogenesis. Am, ameloblast; IEE, inner enamel epithelium; Pre-Am, presecretory ameloblast.
Author Contributions
X. Zhu, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; Z. Ma, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; F. Xie, J. Wang, contributed to conception, design, data analysis and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345231207309 – Supplemental material for ASH2L, Core Subunit of H3K4 Methylation Complex, Regulates Amelogenesis
Supplemental material, sj-docx-1-jdr-10.1177_00220345231207309 for ASH2L, Core Subunit of H3K4 Methylation Complex, Regulates Amelogenesis by X. Zhu, Z. Ma, F. Xie and J. Wang in Journal of Dental Research
Footnotes
Acknowledgements
We thank Prof. Minghan Tong (CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology), who provided the Ash2lflox/flox mice.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (32000571), Shanghai Sailing Program (20YF1423100), Natural Science Foundation of Shanghai (23ZR1437800), and Biological Clinical Sample Project of Shanghai Ninth People’s Hospital (YBKB202222).
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.