
Abstract
Tooth enamel is generated by ameloblasts. Any failure in amelogenesis results in defects in the enamel, a condition known as amelogenesis imperfecta. Here, we report that mice with deficient autophagy in epithelial-derived tissues (K14-Cre;Atg7 F/F and K14-Cre;Atg3 F/F conditional knockout mice) exhibit amelogenesis imperfecta. Micro–computed tomography imaging confirmed that enamel density and thickness were significantly reduced in the teeth of these mice. At the molecular level, ameloblast differentiation was compromised through ectopic accumulation and activation of NRF2, a specific substrate of autophagy. Through bioinformatic analyses, we identified Bcl11b, Dlx3, Klk4, Ltbp3, Nectin1, and Pax9 as candidate genes related to amelogenesis imperfecta and the NRF2-mediated pathway. To investigate the effects of the ectopic NRF2 pathway activation caused by the autophagy deficiency, we analyzed target gene expression and NRF2 binding to the promoter region of candidate target genes and found suppressed gene expression of Bcl11b, Dlx3, Klk4, and Nectin1 but not of Ltbp3 and Pax9. Taken together, our findings indicate that autophagy plays a crucial role in ameloblast differentiation and that its failure results in amelogenesis imperfecta through ectopic NRF2 activation.
Introduction
The epithelial compartment of the developing tooth is called the enamel organ and comprises specific cell types, including inner and outer enamel epithelial cells (Lacruz et al. 2017). The inner enamel epithelial cells differentiate into ameloblasts, which go through presecretory, secretory, transition, and maturation stages. At the presecretory stage, enamel formation begins with an organic protein-enriched matrix deposited on dentin by secretory ameloblasts. During the secretory stage, polarized tall columnar-shaped ameloblasts secrete enamel proteins such as amelogenin, ameloblastin, and enamelin (Bartlett 2013). Once the primary enamel is fully thickened, the secretory ameloblasts enter the transition stage, followed by the maturation stage, in which ameloblasts remove protein and deposit minerals in the enamel matrix (Bartlett 2013). Any failure or abnormality in these steps of amelogenesis results in developmental enamel defects, which recapitulate those seen in amelogenesis imperfecta.
Amelogenesis imperfecta (OMIM #104510) can have autosomal dominant, autosomal recessive, or X-linked inheritance. Based on the distinct phenotype and mode of inheritance, amelogenesis imperfecta can be grouped into 4 major categories: hypoplastic (type I), hypomaturation (type II), hypomineralized/hypocalcified (type III), and hypomature-hypoplastic enamel with taurodontism (type IV) (Aldred et al. 2003). The most common type is hypoplastic, which accounts for 60% to 73% of cases; hypomaturation represents approximately 20% to 40%, and the hypomineralized type accounts for approximately 7% (Chaudhary et al. 2009). All cases typically display discoloration and excessive attrition in addition to their respective characteristics. Healthy rodents have upper incisors coated with brown-color enamel on the labial side and lower incisors with a translucent appearance (Mitsiadis and Luder 2011). In mice, incisors with enamel defects usually have a chalky-white or opaque appearance due to loss of iron ion deposition on the surface of mature enamel, irregular enamel crystals, a missing enamel layer, or a rough enamel surface. The affected molars show flatted cusps or crowns due to the attrition caused by occlusion.
Autophagy is the basic physiological process responsible not only for clearing damaged or unnecessary proteins and organelles from the cells (Dikic and Elazar 2018) but also for supplying amino acids and other nutrients under starvation and stress conditions (Russell et al. 2014). ATG7 (an E1-like enzyme) and ATG3 (an E2-like enzyme) are crucial for the activation of the autophagy pathway (Mizushima and Komatsu 2011). Accumulating evidence indicates that molecules related to autophagy play roles in various nonautophagy cellular functions, including cell differentiation and transcriptional regulation. However, the autophagy-specific molecular mechanisms involved in cell differentiation remain unclear.
Nuclear factor NRF2 (aka NFE2L2), a basic region-leucine zipper-type transcription factor, localizes in the cytoplasm and forms a complex with KEAP1 under normal conditions (Bryan et al. 2013). In the presence of cellular stresses, NRF2 breaks off the binding with KEAP1, which inhibits ubiquitination and degradation of NRF2, accumulates in the cytosol, and then translocates into the nucleus (Bryan et al. 2013), where it binds to the antioxidant response element (ARE; aka NRF2-binding motif) to regulate gene expression (Malhotra et al. 2010). Recent studies show that autophagy regulates NRF2-ARE signaling through p62/SQSTM1, an autophagy adaptor protein. P62/SQSTM1 competes with NRF2 for the interaction with KEAP1 at the NRF2-binding motif (Komatsu et al. 2010; Taguchi et al. 2012). In this study, we investigated the role of autophagy in tooth development.
Materials and Methods
Animals
K14-Cre (Dassule et al. 2000) mice were obtained from The Jackson Laboratory (018964) and used for the generation of epithelial-specific conditional knockout (cKO) mice. Atg7 F/F (Komatsu et al. 2005) and Atg3 F/F (Jia and He 2011) mice were crossed with K14-Cre mice to generate the K14-Cre;Atg7 F/F cKO (Atg7 cKO) and K14-Cre;Atg3 F/F cKO (Atg3 cKO) mutant mice. Atg7 F/F and Atg3 F/F littermate mice were used as wild-type controls. Genotyping was performed by polymerase chain reaction (PCR), as previously described (Suzuki et al. 2019). All mice were bred under pathogen-free conditions with free access to water and food in rooms with a 12-h light/dark cycle. Carbon dioxide inhalation was used for euthanasia. All animal experiments were reviewed and approved by the Animal Welfare Committee and the Institutional Animal Care and Use Committee of UTHealth (AWC-22-0087). Both male and female mice were analyzed at age 8 or 26 wk, with n = 6 per group (from different litters) in each experiment. This study complied with the ARRIVE 2.0 (Animal Research: Reporting of In Vivo Experiments) guidelines for preclinical animal studies.
Cell Culture
The mHAT9d mouse dental epithelial cell line that originated from the apical bud in the incisors, a gift from Dr. Hidemitsu Harada (Iwate Medical University, Iwate, Japan), was cultured as previously described (Otsu et al. 2016). Atg3 knockout (KO) and Atg7 KO mHAT9d cells were generated by the CRISPR/Cas9 method using mouse Atg3 KO and HDR plasmids (sc-426768; Santa Cruz Biotechnology) and mouse Atg7 KO and HDR plasmids (sc-428805; Santa Cruz Biotechnology), according to the manufacturer’s protocol. Mouse Nrf2/Nfe2l2 siRNA (NM-010902; mapping to 2C3, Sc-37049; Santa Cruz Biotechnology) or the negative control siRNA (sc-37007; Santa Cruz Biotechnology) were transfected with Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) into Atg3 and Atg7 KO cells, according to the manufacturer’s protocol. Either the mouse full-length Nrf2/Nfe2l2 cDNA vector (MG56971-U; Sino Biological, Inc.) or the pUC19 backbone vector (50005; addgene) was transfected with Lipofectamine 3000 (Thermo Fisher Scientific) into WT mHAT9d cells, according to the manufacturer’s protocol; 24 h after transfection, the cells were cultured with ameloblast differentiation medium (including 15 µg/mL retinoic acid [R2625, Sigma Aldrich] and 0.1 μM dexamethasone [D4902, Sigma Aldrich]) for 48 h in order to induce ameloblast differentiation.
Micro–Computed Tomography Scanning and 3-Dimensional Reconstruction
The maxillae and mandibles (n = 6 per group) of 8- and 26-wk-old mice were resected and fixed, as previously described (Shim et al. 2022). The micro–computed tomography (micro-CT) scans were performed at a 15-µm resolution with a SCANCO micro-CT-40 system (SCANCO Medical USA, Inc.; 550 peak kilovoltage and 145-µA x-ray source). Three-dimensional (3D) reconstruction and analysis of the micro-CT images from DICOM files were attained with the Dragonfly software (Version 3.6 for Windows; Object Research Systems [ORS], Inc.).
Imaging Analysis for Scanned Incisors and Molars
The region containing enamel in the lower incisor was divided into eight 1-mm-long cross sections (ES1–ES8), starting at 75% of the distance between the opening of the apical foramen and the distal boundary of the last lower molar at the level of the cement–enamel junction. At each segment (ES), a volume of interest (VOI) was created by manually outlining the enamel boundary and excluding nonenamel objects. Each VOI was then used to calculate the average density in Hounsfield units (HUs) and area of enamel (µm2).
Transmission Electron Microscopy
Samples were examined under a JEM1010 transmission electron microscope (JEOL, USA, Inc.), as previously described (Suzuki, Iwaya, et al. 2022). Digital images were obtained with the AMT Imaging System (Advanced Microscopy Techniques Corp.).
Immunoblotting
Ameloblasts were isolated from the upper incisors of Atg7 cKO, Atg3 cKO, and control mice (n = 6 per group) at 8 wk of age or from mHAT9d cells. Immunoblotting was performed as described previously (Suzuki et al. 2020); information on the antibodies used is included in Appendix Table 1.
Histology
Hematoxylin and eosin staining, Berlin blue staining, and immunohistochemistry were performed as previously described (Suzuki et al. 2019); information on the antibodies used is included in Appendix Table 1. Color images were obtained with a light microscope (BX43; Olympus) (n = 6 per group).
Quantitative Reverse Transcription PCR
Total RNAs isolated from the upper and lower incisors of our genetically engineered mouse models or from mHAT9d cells (n = 6 per group) were collected with the QIAshredder and RNeasy mini extraction kit (QIAGEN). Gapdh was used as an internal housekeeping control; the ΔΔ-CT method was applied for the analyses. The PCR primers used in this study are shown in Appendix Table 2.
Comparative Analysis of Transcription Factor Binding Sites
The UCSC genome browser (https://genome.ucsc.edu/) was used to obtain the genomic sequences of the murine genes (Build 38) and 7 additional mammalian genomes, including the 10-kb sequence upstream of the respective transcription start site, as previously described (Suzuki et al. 2019). The multiple alignments were obtained using the Clustal Omega tool with default parameters and settings (Sievers et al. 2011). The ARE, which resembles the NRF2-binding motif (5′-TGAG/CNNNGC-3′) (Suzuki et al. 2019), was searched on the sequence.
Chromatin Immunoprecipitation Assay
Tissue extracts from the maxilla and mandible were incubated with either rabbit polyclonal NRF2 antibody (Invitrogen, PA5-27882) or the standard mouse IgG (Santa Cruz Biotechnology, sc-2025, 1:100). Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Suzuki et al. 2020). There were several binding sites (information about the primers used is provided in Appendix Table 2); the positions of the PCR fragments correspond to NCBI mouse genome Build 38 (mm 10), and n = 6 per group were analyzed in each experiment.
In Situ Hybridization
In situ hybridization for Bcl11b (ACD, 413271), Dlx3 (ACD, 425191), Klk4 (ACD, 483451), and Nectin1 (ACD, 322360) was performed using the RNAscope 2.5 Assay platform (ACD). The color images were obtained with a light microscope (BX43, Olympus), and n = 6 per group were analyzed in each experiment.
Statistical Analysis
All results were obtained from at least 3 independent experiments (n = 6 per group in each experiment). Two-tailed nonparametric Student’s t tests were applied to all experimental analyses. Data were analyzed using the Prism software (GraphPad Software). A P value <0.05 was considered statistically significant. For all graphs, the data are presented as mean ± standard deviation (SD).
Results
Autophagy Deficiency Causes Amelogenesis Imperfecta
To investigate the role of autophagy in tooth development, we analyzed mice with an epithelial tissue-specific deletion of either Atg7 or Atg3 (K14-Cre;Atg7 F/F and K14-Cre;Atg3 F/F mice; hereafter Atg7 cKO and Atg3 cKO mice). Although the upper incisors in wild-type (Atg7 F/F and Atg3 F/F ) and heterozygous (K14-Cre;Atg7F/+ and K14-Cre;Atg3F/+) mice exhibited yellowish pigmentation on the enamel surface, both Atg7 and Atg3 cKO mice exhibited chalky-white upper incisors (Fig. 1A; Appendix Figs. 1A and 2), suggesting that there is a defect in enamel formation in the mutant mice. The molar crown is known to be grinded in amelogenesis imperfecta (Crawford et al. 2007). To confirm the presence of enamel defects in the molars of Atg7 and Atg3 cKO mice, we assessed their shape and surface and found that molar crowns were cracked and flattened (Fig. 1B; Appendix Fig. 1B and 2). Next, to evaluate the mineral density and thickness of tooth enamel, we conducted micro-CT imaging analyses and observed that these parameters were significantly decreased in the lower incisors of Atg7 and Atg3 cKO mice compared to wild-type controls (Fig. 1C, D; Appendix Fig. 2C, D). Similarly, the mineral density and enamel thickness of the molars were decreased in Atg7 and Atg3 cKO mice compared to control mice (Fig. 1E, F; Appendix Fig. 2E, F). Taken together, our results indicate that Atg7 and Atg3 cKO mice exhibit a combination of amelogenesis imperfecta type I (hypoplastic type) and type III (hypomineralized type). Since we detected no difference in phenotypes between Atg7 and Atg3 cKO mice, we concluded that ATG7/ATG3-mediated autophagy plays a crucial role in ameloblast differentiation.
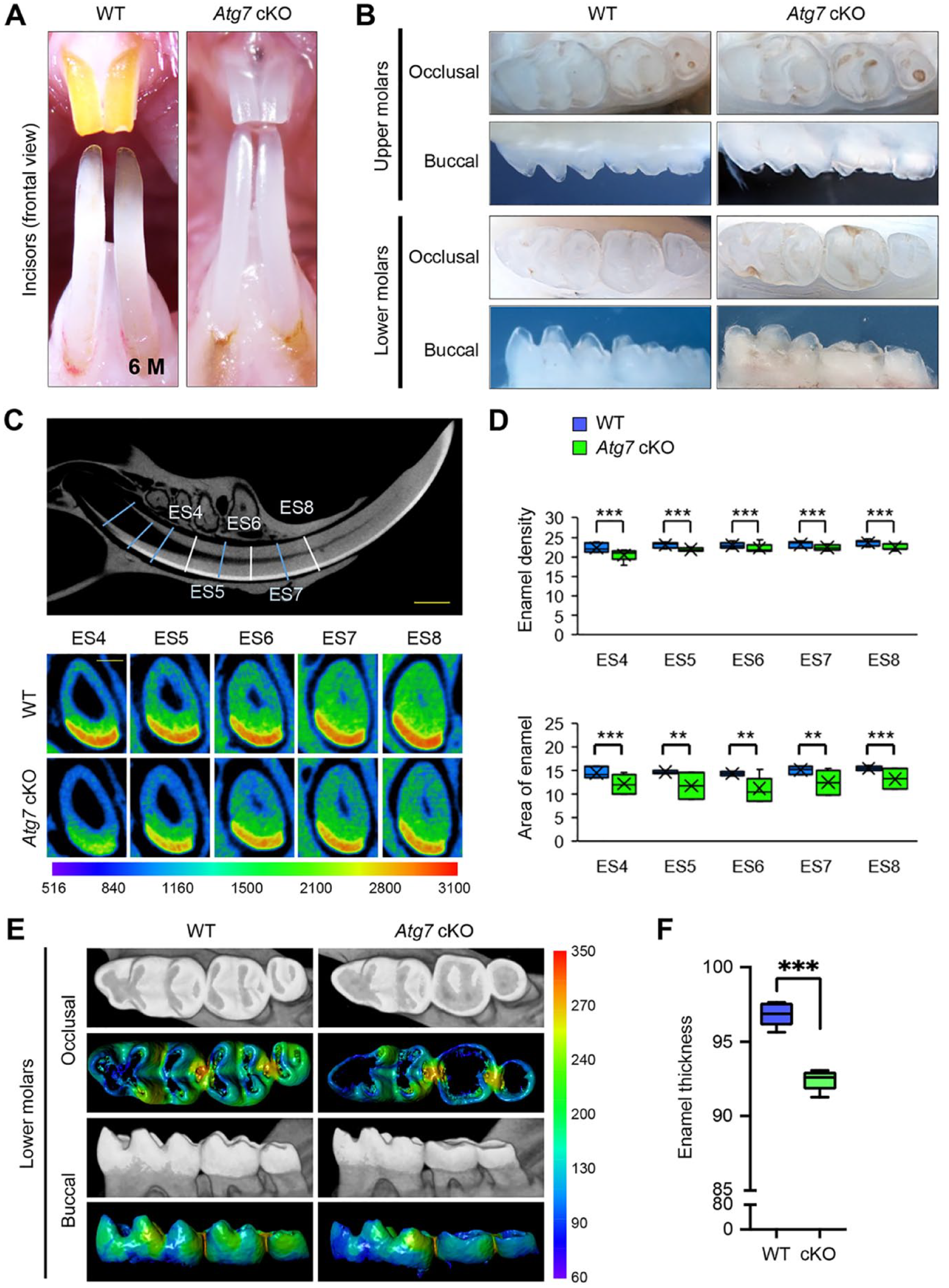
Gross appearance of the teeth of wild-type and Atg7 mutant mice. (
Autophagy Deficiency Results in Failure of Ameloblast Differentiation
To investigate which differentiation stage was affected by loss of autophagy, we conducted histological analyses of the upper and lower incisors of Atg7 cKO, Atg3 cKO, and wild-type control mice at 8 wk of age. The secretory stage of ameloblasts in wild-type mice showed a tall columnar morphology surmounting a thick, well-organized eosinophic enamel extracellular matrix that was indistinguishable in Atg7 and Atg3 cKO mice (Fig. 2A; Appendix Figs. 3 and 4A, B). The morphology of the cells dramatically changes at the transition stage through digestion of superfluous organelles and reorganization of the cytoplasm, suggesting that autophagy may be involved in this reconstruction of cellular structures. As expected, we identified drastic changes in ameloblast morphology at the transition stage (Fig. 2A; Appendix Fig. 4A, B). In addition, we found that ruffle-ended and smooth-ended ameloblasts in Atg7 and Atg3 cKO mice were morphologically similar to the pigment ameloblasts seen in wild-type mice (Fig. 2A; Appendix Fig. 4B). While the cells in wild-type mice at the pigmentation stage (the final stage of ameloblast differentiation) were aligned in a single layer, the cells in Atg7 and Atg3 cKO mice were piled up in multiple layers and presented a round shape (Fig. 2A; Appendix Fig. 4B). The defects in autophagy were confirmed with immunoblotting (Fig. 2B; Appendix Fig. 4C), immunohistochemistry (Fig. 2C; Appendix Fig. 4D), and transmission electron microscopy (Fig. 2D; Appendix Fig. 4E) in incisors of Atg7 and Atg3 cKO mice.
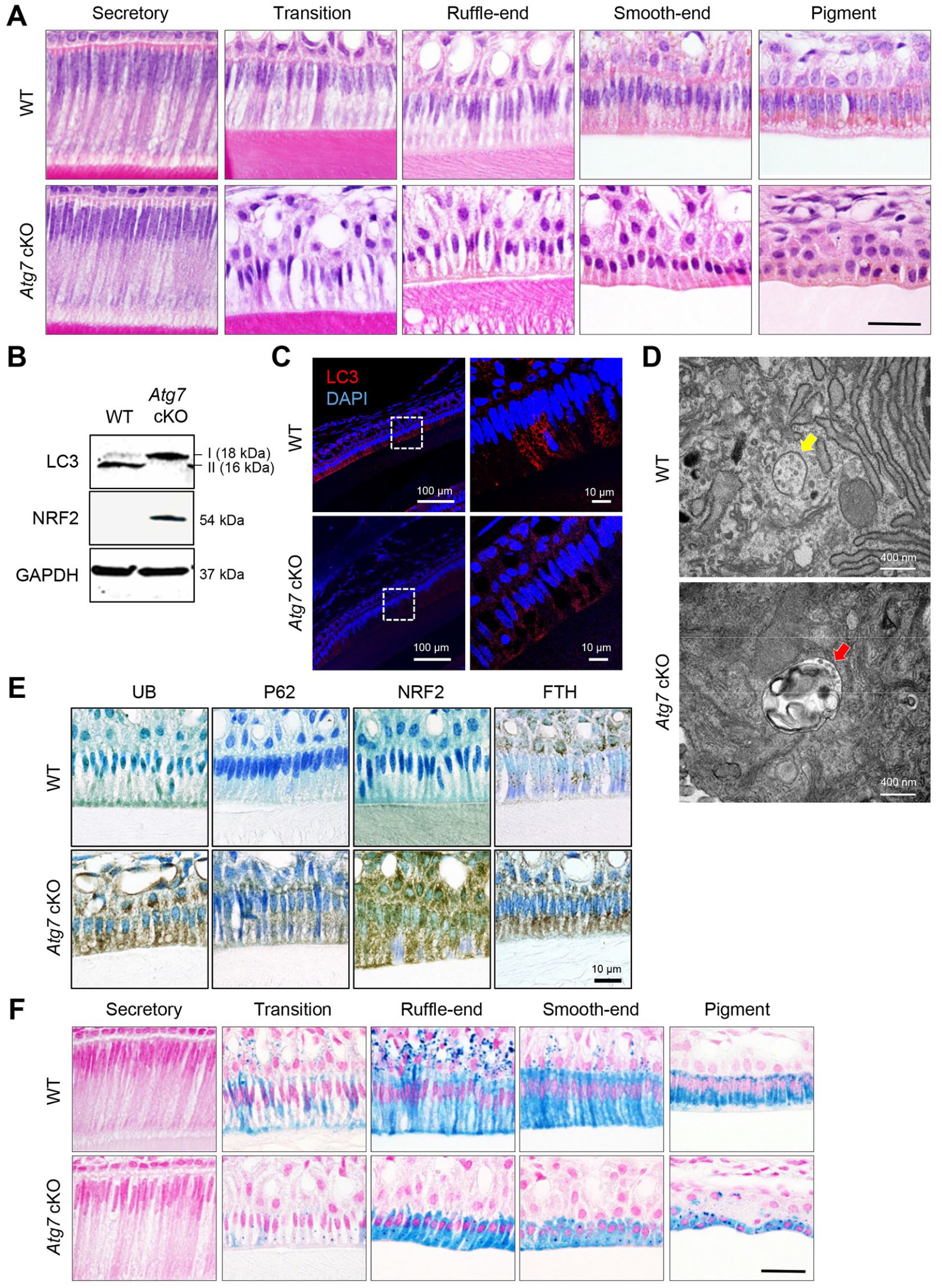
Histological appearance of the incisors of wild-type and Atg7 mutant mice. (
In rodents, iron is deposited onto the mature enamel surface of the incisors, particularly the upper incisors, through the ameloblast maturation to pigmentation stages (Lacruz et al. 2017). We found that ferritin (FTH), a universal cytosolic protein crucial for iron storage and release known to be regulated by NRF2, was abnormally accumulated in ameloblasts in the incisors of Atg7 and Atg3 cKO mice compared to wild-type mice (Fig. 2E; Appendix Fig. 4F). As a result, the iron detected by berlin blue staining was accumulated in ameloblasts after the transition stage in Atg7 and Atg3 cKO mice (Fig. 2F; Appendix Fig. 4G). Taken together, these data show that autophagy is crucial for the transformation of ameloblasts during the secretion-to-maturation stage.
Next, to investigate the molecular mechanism(s) regulating ameloblast differentiation through autophagy, we analyzed the cellular level of autophagy’s substrates (ubiquitinated protein [UB] and p62/SQSTM1) and autophagy-associated transcription factor NRF2. We found that these molecules accumulated in the ameloblasts of Atg7 and Atg3 cKO mice, suggesting that a compromised autophagy process suppresses NRF2 degradation in ameloblasts (Fig. 2E; Appendix Fig. 4F).
Through searches of the Mouse Genome Informatics (MGI) database and the literature, we found 70 genes associated with amelogenesis imperfecta in genetically engineered mice (Suzuki, Yoshioka, et al. 2022). To determine which of these genes were regulated through the autophagic machinery, we analyzed the promoter regions of the candidate genes associated with amelogenesis imperfecta in mice. Among 70 genes, 6 genes (Bcl11b, Dlx3, Klk4, Ltbp3, Nectin1, and Pax9) contained NRF2-binding sites (aka ARE) (Malhotra et al. 2010) in their promotor regions (Fig. 3A; Appendix Fig. 5), and their expression was significantly downregulated in the upper incisors of Atg7 and Atg3 cKO mice compared to wild-type control mice (Fig. 3B; Appendix Fig. 6A). This suggests that these potential amelogenesis imperfecta-associated genes are regulated by the NFR2-ARE pathway through autophagic activity. Next, to test whether these NRF2-binding sites are functionally relevant in the regulation of gene expression, we conducted ChIP assays and found that expression of Bcl11b, Dlx3, Klk4, and Nectin1, but not of Ltbp3 and Pax9, was directly regulated by NRF2 (Fig. 3C; Appendix Fig. 6B). Furthermore, we confirmed that gene and protein expression of Bcl11b, Dlx3, Klk4, and Nectin1 was significantly decreased in ruffle-ended ameloblasts in Atg3 and Atg7 cKO mice compared to wild-type mice (Fig. 4). Importantly, expression of Bcl11b, Dlx3, Klk4, and Nectin1, but not of Ltbp3 and Pax9, was significantly downregulated by Nrf2 overexpression in mouse ameloblast cell line mHAT9d (Fig. 5A). Furthermore, we generated both Atg7 KO and Atg3 KO mHAT9d cells (Fig. 5B, C). As expected, expression of Bcl11b, Dlx3, Klk4, and Nectin1, but not of Ltbp3 and Pax9, was restored by knocking down Nrf2 in Atg7 KO and Atg3 KO mHAT9d cells (Fig. 5D). Taken together, our results indicate that autophagy deficiency results in defects in ameloblast differentiation through NRF2, leading to amelogenesis imperfecta in mice.
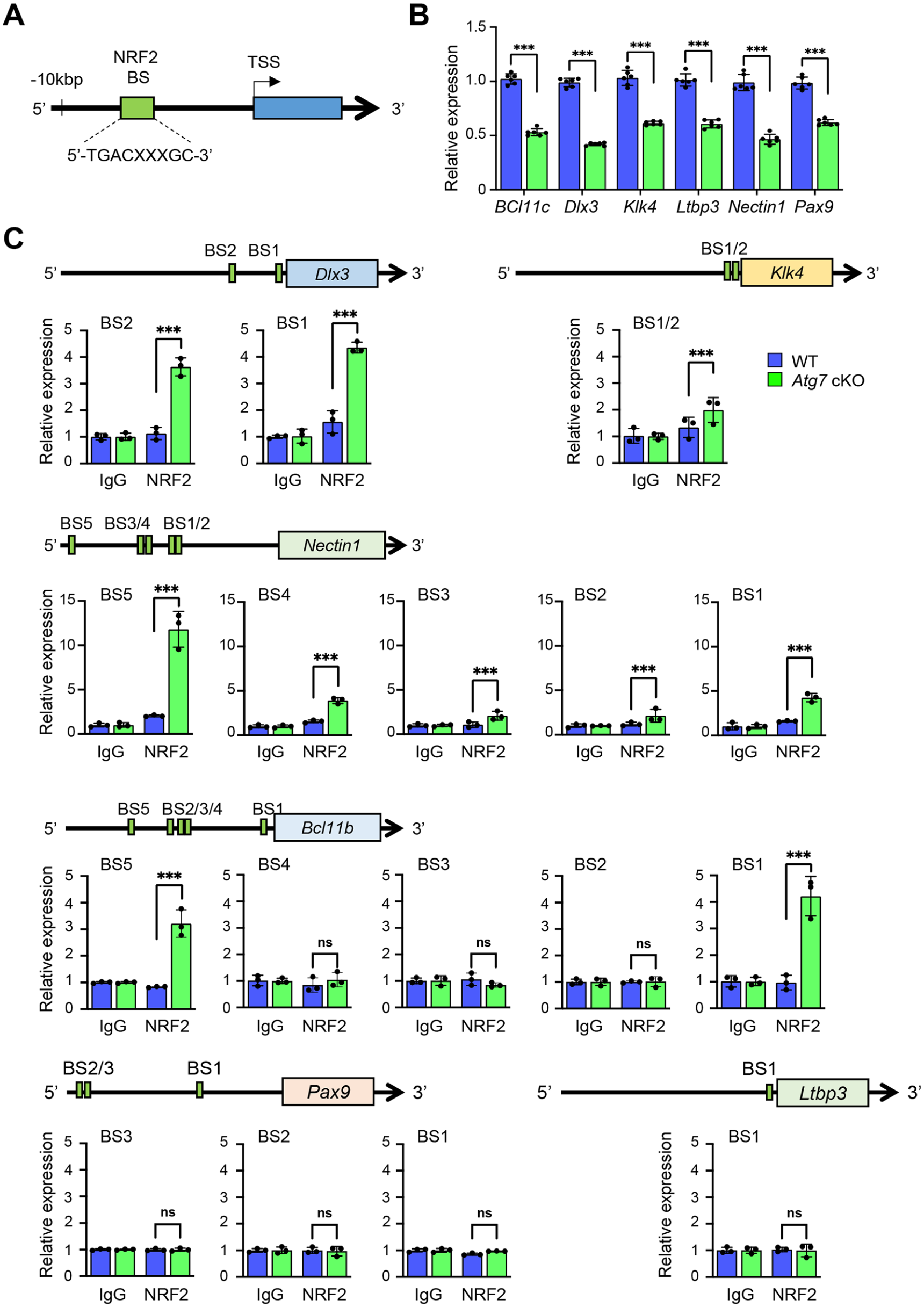
Identification of genes with compromised expression in the teeth of autophagy-deficient mice. (
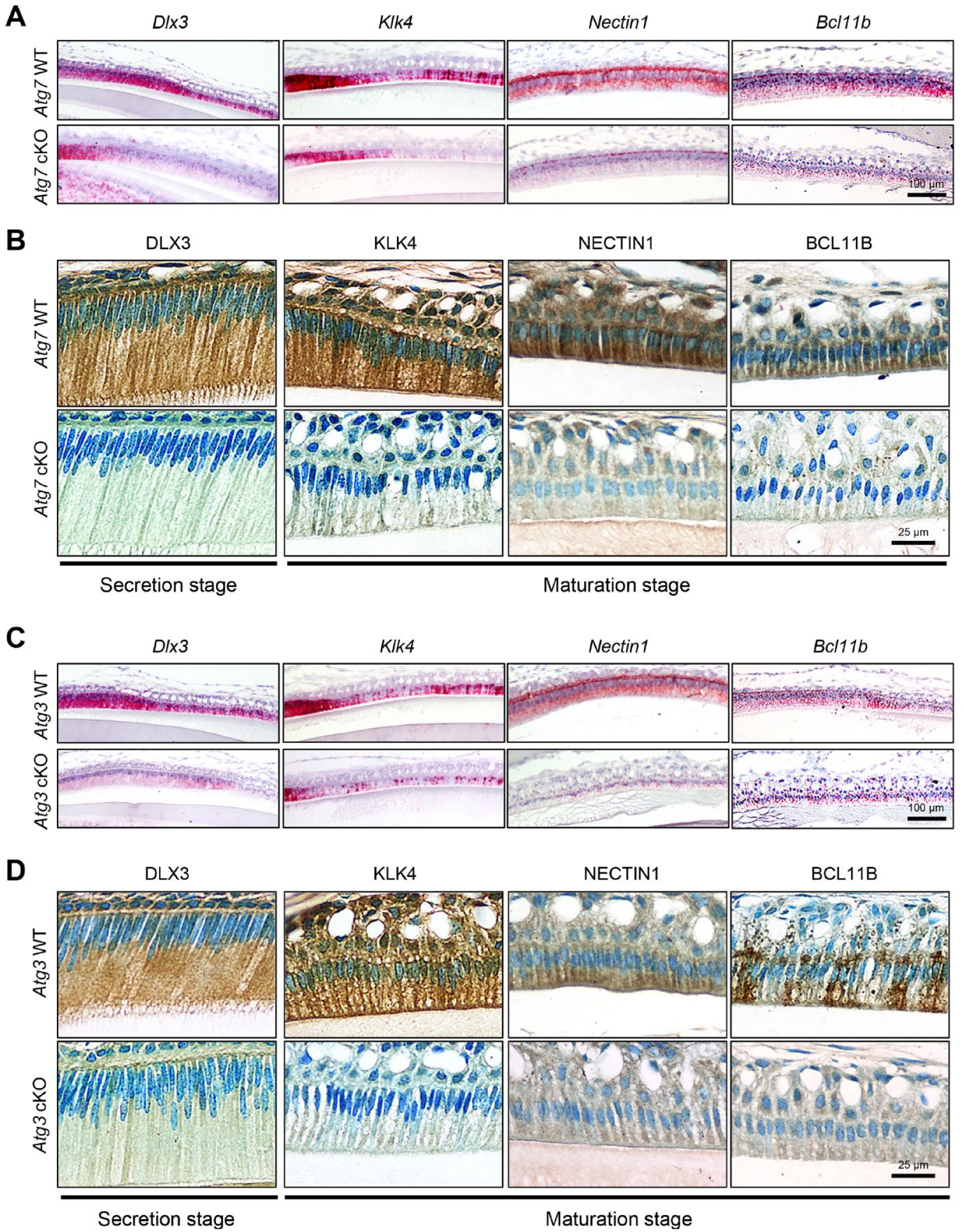
Expression of molecules related to amelogenesis in the upper incisors of autophagy-deficient mice. (
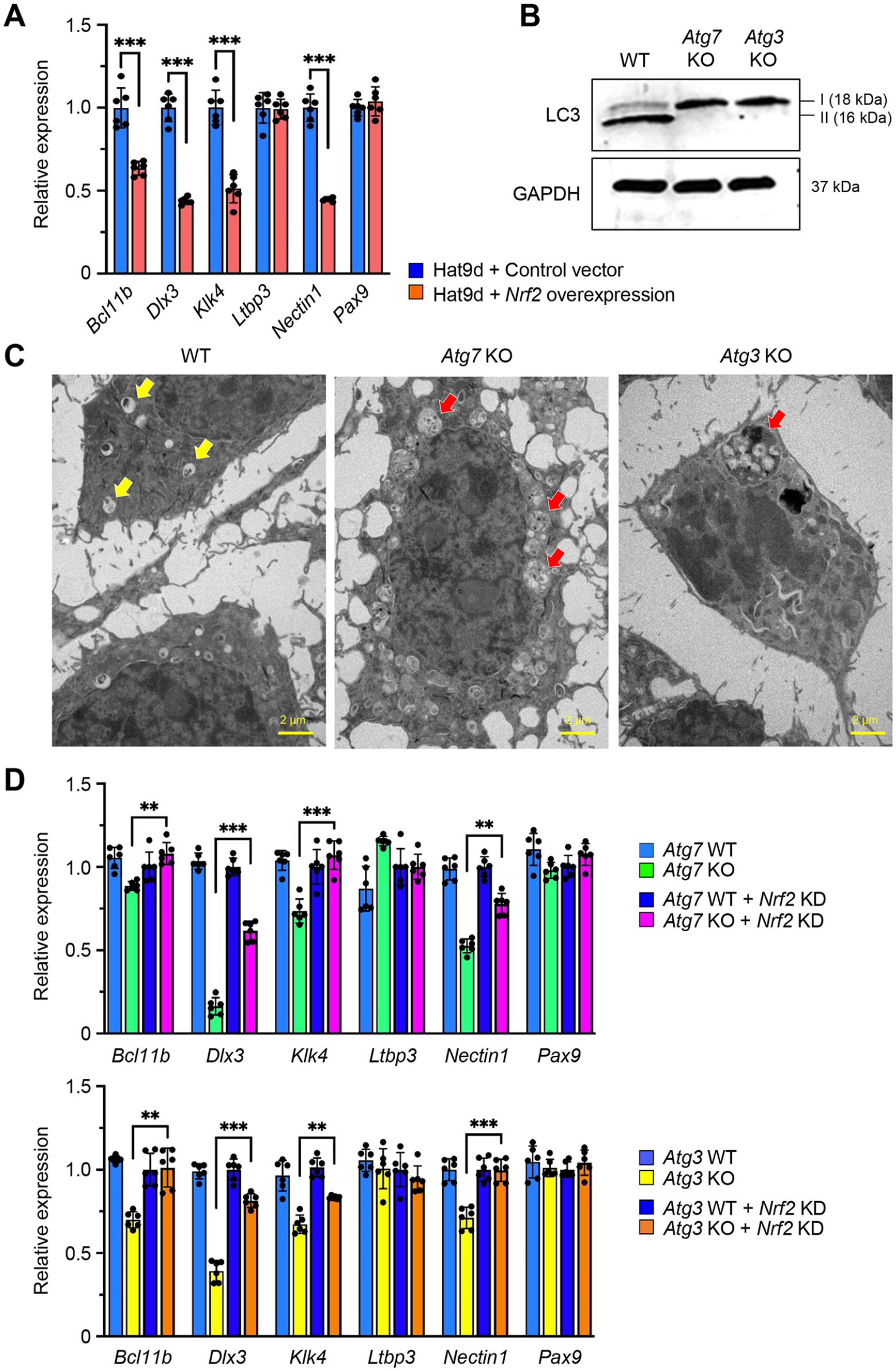
NRF2-dependent regulation of genes in autophagy-deficient ameloblasts. (
Discussion
An increasing number of studies show that autophagy is crucial for protein/organelle degradation as well as various cellular functions, including differentiation and homeostasis (Perrotta et al. 2020). Rodent tooth incisors continuously grow throughout the animal’s lifetime, with ameloblasts lined up in a single line; therefore, they constitute a unique model to investigate the process of ameloblast differentiation in vivo. To evaluate the contribution of autophagy to ameloblast differentiation, we examined the mouse incisors of Atg7 cKO, Atg3 cKO, and control mice and found that expression of Bcl11b, Dlx3, Klk4, and Nectin1 is regulated through NRF2.
BCL11B, a transcriptional repressor, plays critical roles in the development of various tissues and regulates the expression of fibroblast growth factors in the lower incisors (Katsuragi et al. 2013). Mice with a loss of Bcl11b (Bcl11b–/– mice) die at birth and exhibit tooth defects accompanied by failure of ameloblast differentiation (Katsuragi et al. 2013). Moreover, mice with a point mutation (S to G) at position 826 in a Bcl11b heterozygous background (Bcl11bS826G/– mice) display hypoplasia of the upper incisors, but not the lower incisors, due to defects in the production of ameloblast progenitors (Katsuragi et al. 2013). DLX3, a homeodomain-containing transcription factor, is expressed in ameloblasts during the secretory and maturation stages (Duverger et al. 2017). Mice with a deficiency for Dlx3 in enamel organs (K14-Cre;Dlx3 F/F ) display chalky-white incisors with enamel defects (Bartlett and Simmer 2014), and mutations in DLX3 result in tricho-dento-osseous syndrome (TDO, OMIM#190320), a dominantly inherited syndrome characterized by kinky/curly hair at birth, highly mineralized thickened calvaria, and hypoplastic amelogenesis imperfecta with taurodontism (Dong et al. 2005) and nonsyndromic amelogenesis imperfecta of hypoplastic and hypomature types (Bonnet et al. 2020). KLK4 is expressed in ameloblasts during the transition and maturation stages and removes partially hydrolyzed matrix proteins from the enamel layer; mutations in KLK4 result in autosomal recessive, nonsyndromic amelogenesis imperfecta, hypomaturation type, in humans and mice (Hart et al. 2004; Smith et al. 2017). NECTIN (aka PVRL), an immunoglobulin-like cell adhesion molecule, plays a crucial role in cell–cell tight junction formation. Mutations in human NECTIN1 cause ectodermal dysplasia syndromes (Brancati et al. 2010). Mice deficient for Nectin1 (Nectin1–/– mice) exhibit defective enamel formation in their incisors, which are hypomineralized and lack normal iron pigmentation, while the expression pattern of enamel matrix proteins and the protein composition of the enamel matrix are unaffected (Barron et al. 2008).
Although amelogenesis imperfecta may be caused by a combination of multiple factors in humans, this study has a limitation in the analysis of these multifactorial interactions. In addition, although human genetic mutations occur in all cell lineages, in this study, only an ameloblast lineage was analyzed. Future studies may determine the role of cell–cell interactions between the dental epithelium and mesenchyme in amelogenesis imperfecta using various animal models.
In conclusion, this study shows that autophagy plays a crucial role in tooth enamel development, with loss of autophagy leading to amelogenesis imperfecta through ectopic activation of the NRF2 pathway. Our results will help identify novel targets for therapeutics as well as new diagnosis tools for amelogenesis imperfecta and related risk factors.
Author Contributions
C. Iwaya, contributed to design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; A. Suzuki, J. Iwata, contributed to conception and design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; J. Shim, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; C.G. Ambrose, contributed to data acquisition, ally revised the manuscript. All authors provided final approval of the manuscript and agreed to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345231169220 – Supplemental material for Autophagy Plays a Crucial Role in Ameloblast Differentiation
Supplemental material, sj-docx-1-jdr-10.1177_00220345231169220 for Autophagy Plays a Crucial Role in Ameloblast Differentiation by C. Iwaya, A. Suzuki, J. Shim, C. G. Ambrose and J. Iwata in Journal of Dental Research
Supplemental Material
sj-tif-2-jdr-10.1177_00220345231169220 – Supplemental material for Autophagy Plays a Crucial Role in Ameloblast Differentiation
Supplemental material, sj-tif-2-jdr-10.1177_00220345231169220 for Autophagy Plays a Crucial Role in Ameloblast Differentiation by C. Iwaya, A. Suzuki, J. Shim, C. G. Ambrose and J. Iwata in Journal of Dental Research
Footnotes
Acknowledgements
We thank Dr. Masaaki Komatsu (Juntendo University, Tokyo, Japan) and Dr. You-Wen He (Duke University, North Carolina, USA) for providing the Atg7 F/F and Atg3 F/F mice. We also thank Dr. Hidemitsu Harada (Iwate Medical University, Iwate, Japan) for the mHAT9d cells and the High Resolution Electron Microscopy Facility of the MD Anderson Cancer Center.
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Institute of Dental and Craniofacial Research, the National Institutes of Health (R01DE026767 and R01DE029818 to JI), and UTHealth School of Dentistry faculty funding to J. Iwata.
Data and Materials Availability
All data needed to support the conclusions in this article are included in the main text and/or the supplemental materials. Additional data related to this study may be requested from the corresponding author.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.