
Abstract
Ectopic enrichment of oral microbes in the gut is a notable alteration in gut microbial balance. These microbes are likely delivered from the oral cavity with saliva and food; however, evidence of oral–gut microbial transmission is insufficient and needs further investigation. In this observational study, we examined 144 pairs of saliva and stool samples collected from community-dwelling adults to verify the oral–gut microbial link and identify the relevant influencing factors on the increased abundance of oral microbes within the gut. The bacterial composition of each sample was determined using PacBio single-molecule long-read sequencing of the full-length 16S ribosomal RNA gene and amplicon sequence variant (ASV) analysis. Although the bacterial compositions of salivary and gut microbiota were distinctly different, at least 1 ASV was shared between salivary and gut microbiota in 72.9% of subjects. Shared ASVs accounted for 0.0% to 63.1% (median 0.14%) of the gut microbiota in each subject and frequently included abundant Streptococcus salivarius and Streptococcus parasanguinis. Their total relative abundance in the gut was significantly higher in older subjects or those with dental plaque accumulation. The gut microbiota with ≥5% of shared ASVs displayed a higher abundance of Streptococcus, Lactobacillus, and Klebsiella and a lower abundance of Faecalibacterium, Blautia, Megamonas, and Parabacteroides. Our study presents evidence for the translocation of oral bacteria to the gut in community-dwelling adults and suggests that aging and dental plaque accumulation contribute to an increased abundance of oral microbes in the gut, which might be relevant to the compositional shift in the gut commensals.
Introduction
The human intestine is home to numerous microbes that form a diverse and unique microbial community (Human Microbiome Project Consortium 2012; Segata et al. 2012), which is involved in the maintenance of host physiologic homeostasis by contributing to host metabolism and immunity (Round and Mazmanian 2009; Nicholson et al. 2012). In line with this, dysbiosis of gut microbiota is associated with intestinal disorders, including inflammatory bowel disease (IBD) (Franzosa et al. 2019; Lavelle and Sokol 2020) and colorectal cancer (CRC) (Wong and Yu 2019). Recent research indicates that abnormal enrichment of oral taxa is observed in the gut of patients with IBD (Atarashi et al. 2017) and CRC (Mima et al. 2015; Yachida et al. 2019). Although the causal relationship between oral taxa enrichment and intestinal diseases is not fully understood, ectopic colonization of oral taxa in the gut may be relevant to the etiology of intestinal disorders. In fact, intestinal colonization of Klebsiella pneumoniae, originating in saliva from patients with IBD, is reported to induce gut inflammation in genetically susceptible mice (Atarashi et al. 2017).
The oral cavity, the gateway of the digestive tract, also hosts a large microbial community distinct from the gut microbiota (Human Microbiome Project Consortium 2012; Segata et al. 2012). Given that copious amounts of oral microbes are constantly swallowed along with saliva and bolus of food (Humphrey and Williamson 2001), it is reasonable to consider that members of the oral microbiota could be endogenously translocated to the gut, even with antimicrobial barriers such as gastric acidity (Giannella et al. 1972) and bile (Begley et al. 2005). However, whether oral microbiota act as a microbial source for the gut remains to be determined; furthermore, even the detection of oral bacteria in the gut remains controversial (Schmidt et al. 2019; Rashidi et al. 2021; Tanaka et al. 2022). Identification of links between the oral and gut microbiota, factors influencing the increased abundance of oral microbes in the gut, and its impact on gut-indigenous microbiota may provide a novel perspective to understand the relationships among oral microbiota, gut microbiota, and gut microbiota–related diseases.
In this study, we examine paired salivary and stool samples using the PacBio single-molecule long-read sequencing of the full-length 16S ribosomal RNA (rRNA) gene along with the amplicon sequence variant (ASV) approach. Rapidly advanced PacBio long-read sequencing in the past several years has provided highly accurate sequences using the circular consensus sequencing (CCS) mode, which repeatedly determines the template DNA sequences based on long sequencing potential (Wenger et al. 2019). This sequencing technology can accurately analyze the full-length 16S rRNA gene containing 9 hypervariable regions, enabling taxonomic discrimination at high resolution (Kaan and Zaura 2022, Kageyama et al. 2022). The ASV approach, which resolves differences of as little as 1 nucleotide, can further enhance the taxonomic resolution to the strain level. This study precisely evaluates the oral–gut microbial links using these approaches and identifies the factors influencing the increased abundance of oral microbes in the gut.
Materials and Methods
Study Subjects
The subjects of this observational study were community-dwelling adults in Hisayama town, Japan (Hata et al. 2013). We conducted dental examinations and saliva sampling as a part of a health examination for Hisayama residents in 2017. We also collected stool samples from Hisayama residents who participated in a health examination conducted in 2018. In total, 2,642 participants underwent both dental examination and saliva sampling, of whom 1,362 also participated in stool sampling. We reduced the sample size due to budget constraints and sampled 146 subjects from each age group in 10-y increments (40, 50, 60, 70, or 80 y old) to ensure broad age coverage. Only age was used for the selection of subjects. After excluding 2 subjects with insufficient samples, 144 pairs of saliva and stool samples were examined. Written informed consent was obtained from all the participants. The ethics committee of Kyushu University approved the present study and procedure for obtaining informed consent (approval number: 2021-96 and 743-02).
Health Information and Sample Collection
The oral and general health information obtained in 2017, including age, sex, number of present teeth, sites with probing depth ≥4 mm, bleeding on probing (BOP), decayed teeth, dental plaque, body mass index (BMI), hypertension, diabetes, smoking habits, and alcohol intake, was considered for analysis. Details of the oral and general health information and saliva and stool sampling are described in the Appendix Methods.
DNA Extraction, Sequencing, and Data Processing
DNA was extracted from the saliva samples using the bead-beating method described previously (Kageyama et al. 2022) after the supernatant was discarded following centrifugation. From stool samples, DNA was extracted using an automated DNA isolation system (GENE STAR PI-480; Kurabo Industries Ltd.) following bead-beating homogenization. The full-length 16S rRNA gene amplicon analysis was performed using the PacBio Sequel II/IIe system (Pacific Biosciences) and DADA2 pipeline (version 1.14.0) software (Callahan et al. 2016), as previously described (Kageyama et al. 2022) (Appendix Methods). The taxonomy of each sequence variant was first determined using the RDP classifier, with a minimum support threshold of 80%, and the RDP taxonomic nomenclature (to the genus level) (Wang et al. 2007). Next, the species-level taxonomic assignment was performed using BLAST (Altschul et al. 1990) against eHOMD version 15.22 (Chen et al. 2010). Nearest-neighbor species with ≥98.5% identity were selected as candidates for each sequence. Finally, the species-level taxonomy of sequences without hits against the eHOMD was further determined using assignTaxonomy function in DADA2 using silva_nr99_v138_wSpecies_train_set.fa.gz and bootstrap confidence of 80% (Quast et al. 2013; Yilmaz et al. 2014; Callahan et al. 2016).
Statistical Analysis
All statistical analyses were performed using the R software. The differences in bacterial composition between salivary and gut microbiota were evaluated using Bray–Curtis distance based on the ASV abundance data. A phylogenetic tree was constructed based on the sequences of the most abundant ASV in each genus using the ape package in R (Paradis and Schliep 2019). In this study, we defined ASVs detected in both salivary and gut microbiota as shared ASVs for each subject and calculated the total abundance of the shared ASVs in each subject. To evaluate oral-to-gut transmissibility, we calculated the transfer ratio for each genus and species, which was the percentage of subjects with identical ASVs in the gut and saliva for each genus and species found in saliva. The total abundance of the shared ASVs in the gut according to each clinical factor was compared using the Kruskal–Wallis test, along with the calculation of effect size (η2), and again using the Steel–Dwass test. The differentially abundant genera were identified using linear discriminant analysis effect size (LEfSe) between subjects with and without ≥5% relative abundance of the shared ASVs in gut (Segata et al. 2011). The relative abundance of predominant genera was compared using the Mann–Whitney U test.
Results
Characteristics of Subjects and Full-Length 16S rRNA Gene Sequencing
We examined 144 pairs of saliva and stool samples collected from residents aged 40, 50, 60, 70, or 80 y in Hisayama, Japan. Detailed characteristics of the subjects are presented in Appendix Table 1. Most subjects (86.2%) had 20 or more teeth, and half of them (43.1%) had 28 teeth. More than half (58.3%) of the subjects had neither sites with probing depth ≥4 mm nor decayed teeth. However, visibly detectable dental plaque on the tooth surface was observed in 79.9% of subjects, and moderate accumulation was observed in 24.3% of subjects. The intraclass correlation used as a measure of interexaminer reproducibility was 0.747 for probing depth. A total of 288 samples from 144 subjects were analyzed using full-length 16S rRNA gene amplicon analysis with PacBio long-read sequencing. Finally, 1,377,489 denoised reads (6,153.8 ± 6,962.0 reads per saliva sample and 3,412.1 ± 2,858.7 reads per stool sample) and 10,065 ASVs were obtained.
Microbial Difference and Link between Salivary and Gut Microbiota
First, we compared the overall bacterial composition of the salivary and gut microbiota. According to the principal coordinate analysis (PCoA) plot based on the Bray–Curtis distance at the ASV level, the bacterial compositions in saliva and stool were distinctly different (Fig. 1A). In the gut microbiota, 16 genera were identified as predominant genera with ≥1% mean relative abundance. Especially Bacteroides and Faecalibacterium showed higher abundance (Fig. 1B). The salivary microbiota was dominated by 10 genera, particularly Streptococcus and Neisseria, and differed significantly from the gut microbiota. In contrast, Streptococcus and Prevotella were predominant in both, and Veillonella and Megasphaera overlapped slightly. Surprisingly, the distance between the paired samples was 1.0 in only 39 subjects (27.1%), which indicated no ASV sharing and completely different microbial communities in the saliva and gut (Fig. 1C). In comparison, the paired samples from 105 subjects (72.9%) demonstrated a distance of <1.0, indicating that at least 1 ASV was shared between salivary and gut microbiota.
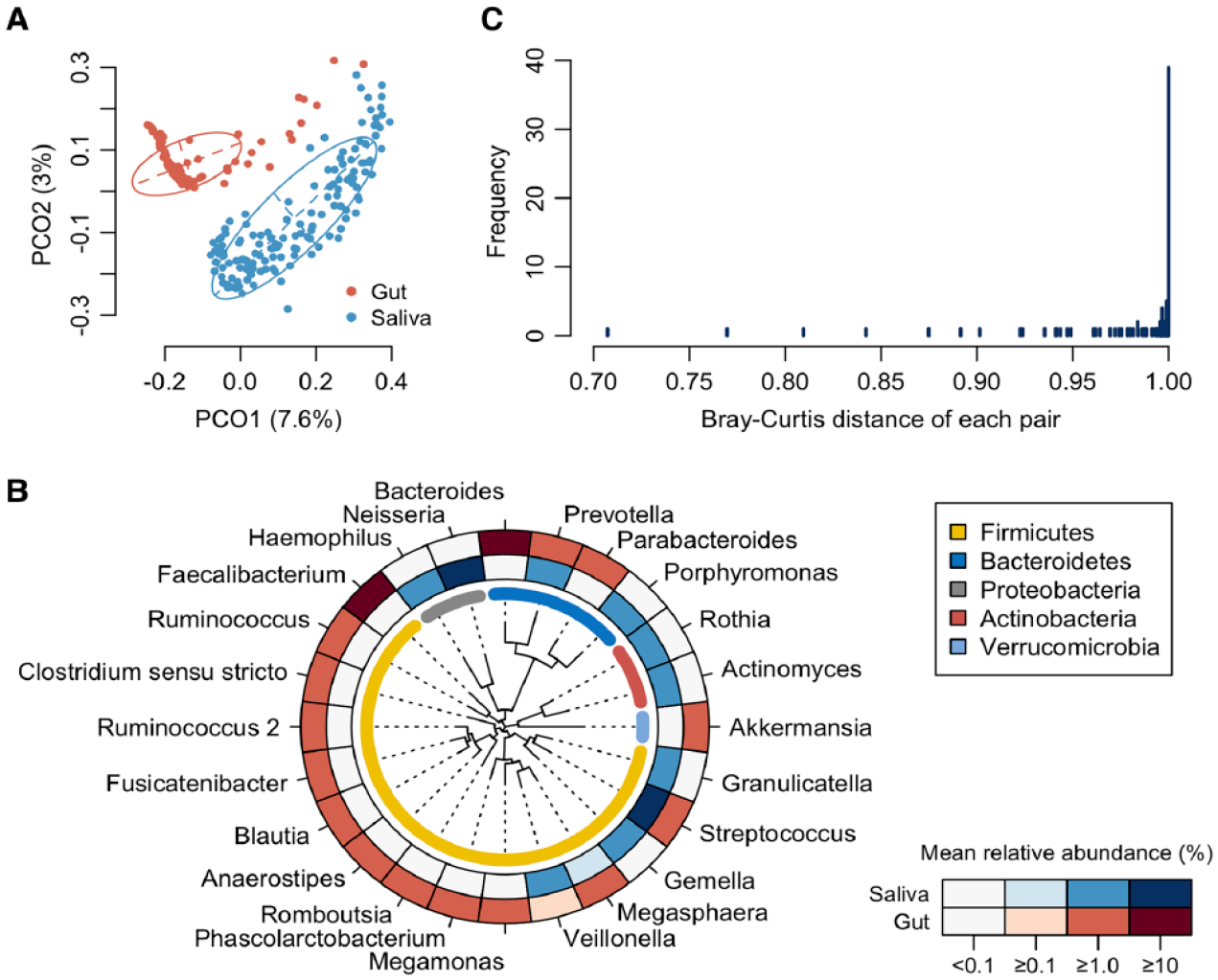
Comparison of salivary and gut microbiota composition. (
To evaluate the extent of microbial sharing between the saliva and gut, we calculated the occupancy of salivary ASVs in the gut microbiota and the sharing frequency of the major salivary bacteria. Of the ASVs detected in the gut microbiota, ASVs shared with their own salivary microbiota were defined as the shared ASVs for each subject. As implicated from the results shown in Figure 1C, the shared ASVs were identified in the gut microbiota of 105 subjects (72.9%) and accounted for a 0.14% median relative abundance (range 0.0%–63.1%) of their gut microbiota (Fig. 2A). The shared ASVs in the gut were predominantly Streptococcus salivarius HMT-755, Streptococcus parasanguinis HMT-411, and Veillonella dispar HMT-160 (Fig. 2B). According to the transfer ratio, Streptococcus was the most frequently shared among the predominant genera in saliva (transfer ratio 0.681, Fig. 2C). Out of the 144 subjects with ASVs corresponding to Streptococcus in salivary microbiota, identical ASVs were detected in the gut microbiota of 119 subjects (68.1%). Although the genus Prevotella was detected in 143 salivary and 45 stool samples, the transfer ratio was 0.0, and there was no ASV sharing. The genus Megasphaera also showed a transfer ratio of 0.0. At the species level, S. salivarius and S. parasanguinis exhibited frequent ASV sharing (transfer ratios of 0.587 and 0.274, respectively). Interestingly, the transfer ratio of each genus and species was not consistent with their relative abundances in the salivary microbiota (Fig. 2C). For instance, the genus Neisseria was the second-most predominant in saliva but had a transfer ratio of 0.007 (only one sharing among 136 subjects with Neisseria in saliva).
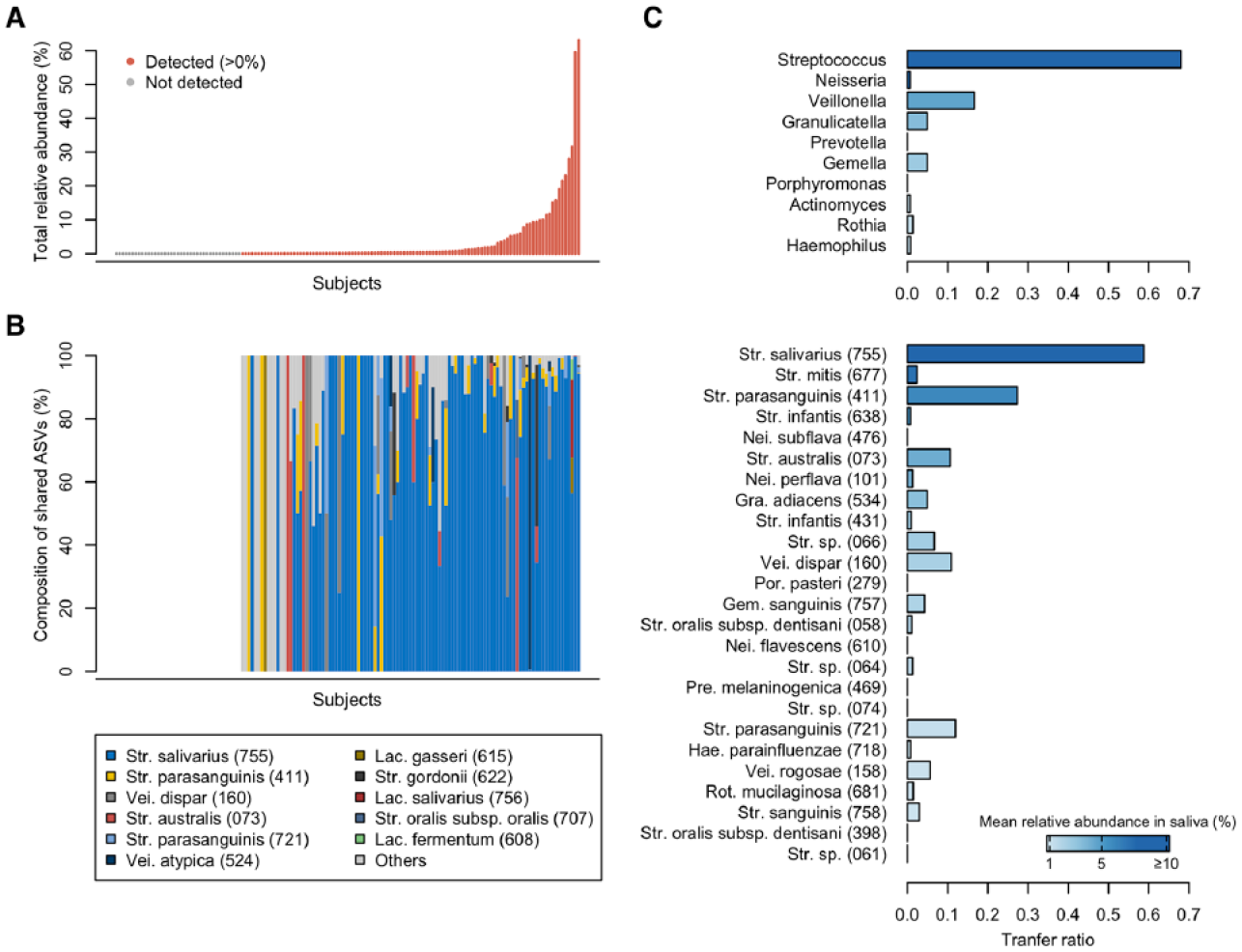
Microbial sharing between salivary and gut microbiota. (
Oral–Gut Microbial Sharing within an Individual
Next, we evaluated microbial sharing and compositional similarity between salivary and gut microbiota within an individual (intrapair) compared to those across different individuals (interpair). The relative abundance and observed number of shared ASVs in gut microbiota in the intrapairs were higher than or equal to those in the interpairs, except for that in 3 subjects and 1 subject, respectively (Appendix Fig. 1A, B). The similarity of bacterial composition in intrapairs was also significantly higher than that in interpairs (Appendix Fig. 1C).
Influential Factors on the Increased Abundance of Oral Bacteria in the Gut
To explore the clinical factors affecting the increased abundance of oral bacteria in the gut, we compared the occupancy of salivary ASVs in the gut microbiota according to clinical characteristics. The Kruskal–Wallis test demonstrated that age and dental plaque score were significantly associated with the total relative abundance of shared ASVs in the gut (Fig. 3A). The abundance showed an increasing trend with age and was significantly higher in 80-y-old subjects than in 40- and 50-y-olds (Fig. 3B). Similarly, subjects with a moderate accumulation of dental plaque had a higher abundance of shared ASVs in the gut than subjects with no dental plaque. There were no significant differences in abundance according to sex, number of present teeth, ≥4 mm probing depth, BOP, decayed teeth, BMI, hypertension, diabetes, smoking habits, or alcohol intake (Fig. 3A).
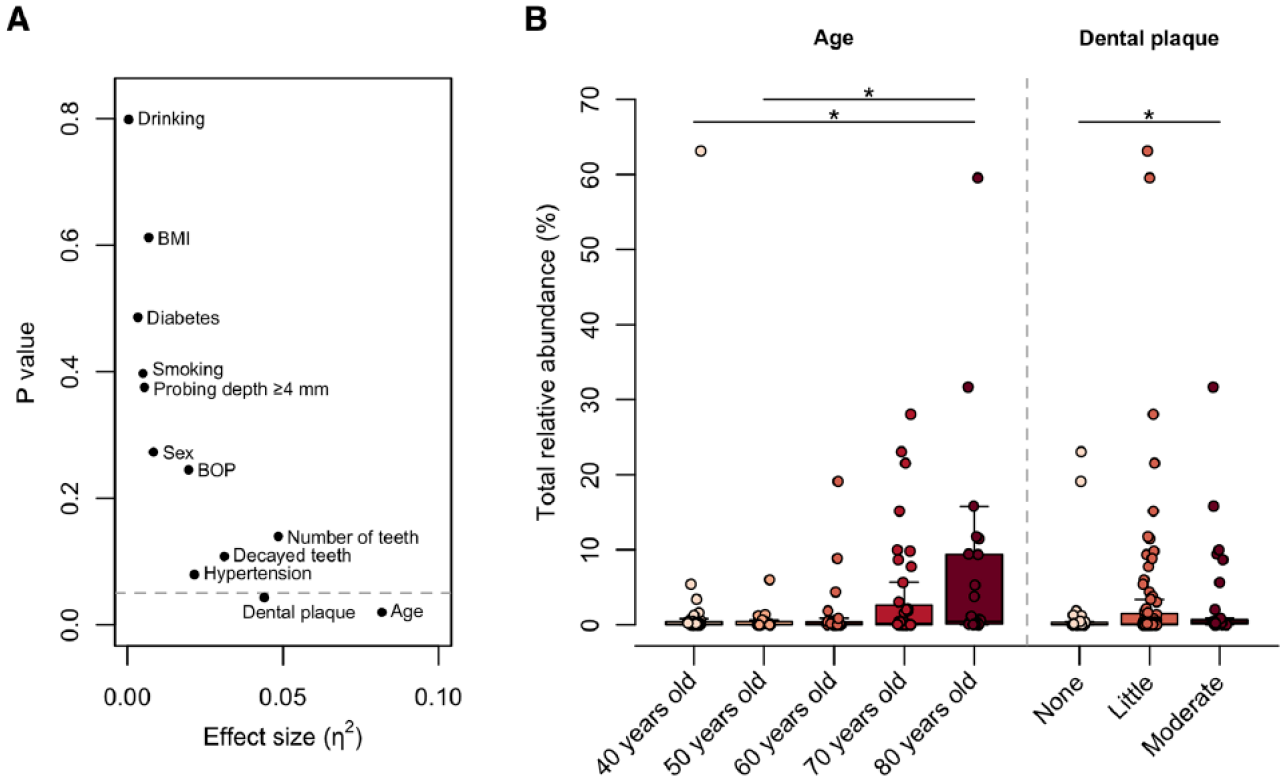
Factors influencing the increased abundance of oral bacteria in the gut. (
Gut Microbiota Composition Associated with Enrichment of Oral Bacteria
Finally, we examined the association between the enrichment of salivary ASVs and the bacterial composition of the gut microbiota. According to the PCoA plot, where the gradient color expresses the total abundance of shared ASVs, the gut microbiota with a higher abundance of shared ASVs was naturally more similar to the salivary microbiota (Appendix Fig. 2). The LEfSe approach identified 7 discriminant genera between subjects with ≥5% (n = 22) and <5% (n = 122) relative abundances of shared ASVs in the gut (Fig. 4A). In subjects with higher shared ASVs in the gut, the relative abundances of Streptococcus, Lactobacillus, and Klebsiella in the gut microbiota were significantly increased (Fig. 4B). In contrast, the abundances of Faecalibacterium, Blautia, Megamonas, and Parabacteroides were significantly lower in the gut. Their differences were mainly attributed to significantly higher abundances of S. salivarius, S. parasanguinis, Lactobacillus salivarius HMT-756, and K. pneumoniae HMT-731 and significantly lower abundances of Faecalibacterium prausnitzii and Parabacteroides distasonis (Appendix Fig. 3). Between subjects with ≥5% and <5% of shared ASVs, there were no differences in the number of observed ASVs (median of 52 and 60 ASVs, respectively) and Shannon index (median of 3.25 and 3.33, respectively) in their gut microbiota.
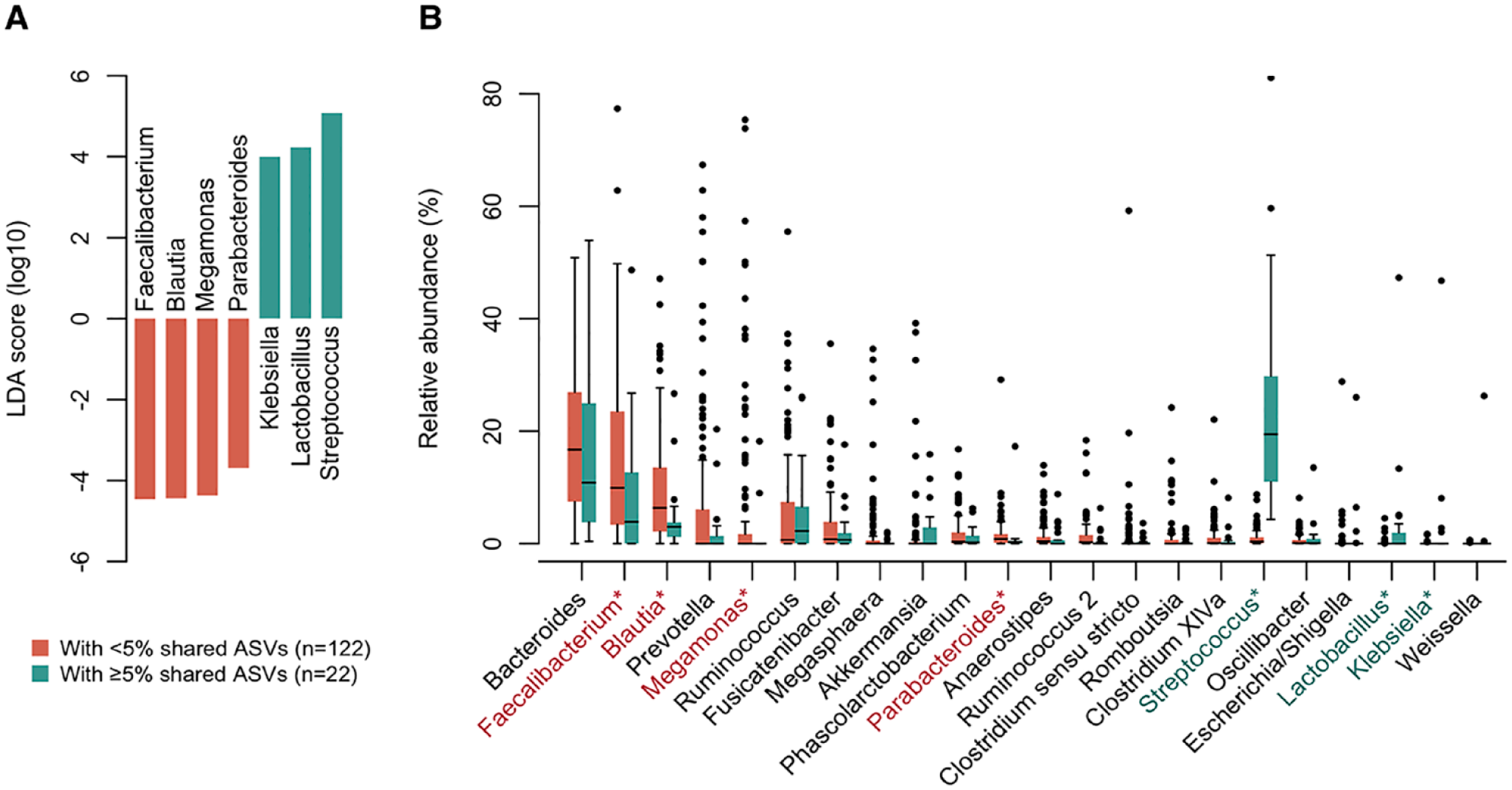
Gut microbiota composition associated with enrichment of oral bacteria. (
Discussion
This study examined 144 pairs of saliva and stool samples using full-length 16S rRNA gene amplicon analysis and the ASV approach and verified the translocation of oral bacteria to the gut based on the ASV sharing (i.e., identical match of full-length 16S rRNA gene). The ASVs shared with own salivary microbiota were detected from gut microbiota in 72.9% of subjects and accounted for 0.0% to 63.1% of gut microbiota for each subject (Fig. 2A). These results demonstrate that translocation of oral bacteria into the gut occurs extensively in community-dwelling adults. This observation supports the findings of a previous study that suggested the oral–gut transmission of microbes in healthy subjects using a metagenomic approach (Schmidt et al. 2019). Further, the oral–gut microbial sharing and compositional similarity were higher within an individual compared to those across different individuals (Appendix Fig. 1). Although the process of translocation was not examined in this study, these findings suggest that oral bacteria in the gut endogenously originate from their oral cavity.
Interestingly, aging was significantly associated with an increased abundance of shared ASVs in the gut microbiota (Fig. 3B). Considering the decline in barrier function due to aging alterations in the gastrointestinal tract (Drozdowski and Thomson 2006), an increase in oral bacteria in the gut is not surprising. Importantly, enrichment was observed in community-dwelling adults who visited our health examination and not in a specific disease population. Although the difficulty lies in distinguishing between pathologic alterations and aging-induced changes, the translocation of oral bacteria to the gut can be considered an age-related change in humans that occurs widely in elderly individuals. Meanwhile, the accumulation of dental plaque was also associated with higher levels of shared ASVs in the gut (median of 0.04%, 0.10%, and 0.31% for each group, Fig. 3B). Although possible confounding due to unmeasured factors is not excluded, this result implies that poor oral hygiene status, including dental plaque accumulation, may contribute to the enrichment of oral bacteria in the gut owing to an increase in ingested bacteria. Oral care may help to prevent the translocation of oral bacteria into the gut. The influence of oral care on oral–gut translocation needs to be further investigated in an interventional study of oral hygiene.
In the subjects with enriched (≥5%) shared ASVs in their gut, decreases of some known gut commensals such as Faecalibacterium, Blautia, Megamonas, and Parabacteroides were observed, not only increases of oral bacteria. A similar result was also obtained in a sensitivity analysis according to 3 categories (with ≥5%, with <5%, and no detection of shared ASVs) (Appendix Fig. 4). At the species level, in subjects with enriched shared ASVs in the gut, the relative abundances of Faecalibacterium prausnitzii and Parabacteroides distasonis were particularly low compared to subjects with <5% of shared ASVs (Appendix Fig. 3). Owing to their ability of high butyrate production, F. prausnitzii is a well-known anti-inflammatory gut commensal bacterium (Sokol et al. 2008) and P. distasonis was also reported to have anti-inflammatory properties (Cekanaviciute et al. 2017). Furthermore, although most ASVs were not assigned to species-level taxa, the genus Blautia was reportedly associated with a low level of visceral fat area (Ozato et al. 2019) (Fig. 4, Appendix Fig. 3). In contrast, in addition to S. salivarius, S. parasanguinis, and L. salivarius that were abundantly detected as the shared ASVs, the abundance of K. pneumoniae, known to be associated with gut inflammation in mice, was also significantly higher in their gut (Atarashi et al. 2017; Kitamoto et al. 2020). Although the causal association was unexplained, these results suggest that the enrichment of oral bacteria in the gut is associated with a decrease in beneficial and an increase in pathogenic bacteria.
This study has some limitations. First, sample collections of saliva and stool were conducted with about a 1-y gap (the average gap was 11.7 ± 1.2 mo). Although salivary microbiota are comparatively steady in microbial communities of the human body (Zhou et al. 2013), this gap potentially leads to an underestimation of the relative abundance and number of shared ASVs in the gut due to the possible annual variability in salivary microbiota. Second, read number differences in each sample may also facilitate the underestimation of microbial sharing. Third, the convenient sampling considering only age of this study could introduce bias. However, there were no significant differences in oral and general health conditions between included and excluded participants (Appendix Table 2). Fourth, 16S rRNA gene analysis cannot evaluate whether salivary bacteria in the gut are alive or dead. However, considering their dominance in the gut, it is unlikely that ingested salivary bacteria constitute a large part of the gut microbiota without growing in the gut. A previous report using a culture approach demonstrated that F. nucleatum strains identical to oral strains were isolated alive from the gut (Komiya et al. 2019). Fifth, the causal association between oral bacteria enrichment and the gut compositional shift remains unexplained; thus, future studies should further examine the role of translocated oral bacteria and their interactions with the gut microbiota.
In conclusion, this study combined full-length 16S rRNA gene amplicon analysis and the ASV approach and demonstrated the translocation of oral bacteria into the gut in community-dwelling adults with high resolution. The occupancy of ASVs shared with the oral cavity was significantly higher in older subjects, suggesting that the enrichment of oral bacteria is an age-related alteration in humans. Although the causal association was unexplained, the enrichment was relevant to the compositional shift in gut-indigenous microbiota with a decrease in known beneficial bacteria. Furthermore, subjects with dental plaque accumulation showed an increased abundance of salivary ASVs in the gut. This result suggests that the translocation of oral bacteria to the gut may be prevented by improving oral hygiene status. Further analyses should be performed to determine the association between ectopically detected oral bacteria and gut microbiota or the subsequent risk of diseases.
Author Contributions
S. Kageyama, contributed to conception and design, data analysis, drafted and critically revised the manuscript; S. Sakata, T. Takeshita, M. Furuta, T. Ninomiya, Y. Yamashita, contributed to conception and design, data analysis, critically revised the manuscript; J. Ma, M. Asakawa, contributed to data analysis, critically revised the manuscript. All authors gave their final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345231160747 – Supplemental material for High-Resolution Detection of Translocation of Oral Bacteria to the Gut
Supplemental material, sj-docx-1-jdr-10.1177_00220345231160747 for High-Resolution Detection of Translocation of Oral Bacteria to the Gut by S. Kageyama, S. Sakata, J. Ma, M. Asakawa, T. Takeshita, M. Furuta, T. Ninomiya and Y. Yamashita in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by JSPS KAKENHI grant numbers JP22K17269, JP20K18808, JP22H03303, JP20K21682, JP20H 03901 and JP21H03200.
Data Availability
The sequence data have been deposited in the DDBJ Sequence Read Archive under accession number DRA015490.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.