
Abstract
Recent genome-wide association studies have suggested novel risk loci associated with periodontitis, which is initiated by dysbiosis in subgingival plaque and leads to destruction of teeth-supporting structures. One such genetic locus was the tumor necrosis factor receptor–associated factor 3 interacting protein 2 (TRAF3IP2), a gene encoding the gate-keeping interleukin (IL)–17 receptor adaptor. In this study, we first determined that carriers of the lead exonic variant rs13190932 within the TRAF3IP2 locus combined with a high plaque microbial burden was associated with more severe periodontitis than noncarriers. We then demonstrated that TRAF3IP2 is essential in the IL-17–mediated CCL2 and IL-8 chemokine production in primary gingival epithelial cells. Further analysis suggested that rs13190932 may serve a surrogate variant for a genuine loss-of-function variant rs33980500 within the same gene. Traf3ip2 null mice (Traf3ip2–/–) were more susceptible than wild-type (WT) mice to the Porphyromonas gingivalis–induced periodontal alveolar bone loss. Such bone loss was associated with a delayed P. gingivalis clearance and an attenuated neutrophil recruitment in the gingiva of Traf3ip2–/– mice. Transcriptomic data showed decreased expression of antimicrobial genes, including Lcn2, S100a8, and Defb1, in the Traf3ip2–/– mouse gingiva in comparison to WT mice prior to or upon P. gingivalis oral challenge. Further 16S ribosomal RNA sequencing analysis identified a distinct microbial community in the Traf3ip2–/– mouse oral plaque, which was featured by a reduced microbial diversity and an overabundance of Streptococcus genus bacteria. More P. gingivalis was observed in the Traf3ip2–/– mouse gingiva than WT control animals in a ligature-promoted P. gingivalis invasion model. In agreement, neutrophil depletion resulted in more local gingival tissue invasion by P. gingivalis. Thus, we identified a homeostatic IL-17-TRAF3IP2-neutrophil axis underpinning host defense against a keystone periodontal pathogen.
Introduction
Periodontitis, as a complex trait disease, is associated with risk alleles with varying effect sizes that have been reported by several genome-wide association (GWA) studies (Divaris et al. 2013; Offenbacher et al. 2016). Through a gene-set enrichment GWA analysis in the Atherosclerosis Risk in Communities (ARIC) participants, we recently reported several genes in which the combined overall single-nucleotide polymorphism (SNP) variants were significantly associated with several periodontal complex traits (PCTs) defined by clinical disease classification and biological intermediates, including microbial and inflammatory characteristics (Offenbacher et al. 2016). For example, the combined SNP variants across the tumor necrosis factor receptor–associated factor 3 interacting protein 2 (TRAF3IP2, ACT1, or CIKS) gene were significantly associated with PCT3, which was characterized by a high loading of gingival crevicular fluid interleukin (IL)–1β level and Aggregatibacter actinomycetemcomitans in subgingival plaque (Offenbacher et al. 2016).
TRAF3IP2 encodes a nonredundant adaptor protein for the IL-17 receptor (IL-17R) that mediates IL-17 responses in several cell types (Huang et al. 2007; Boisson et al. 2013; Ha et al. 2014; Wu et al. 2014). IL-17 family members, including IL-17(A), IL-17E (IL-25), and IL-17F, rely on TRAF3IP2 to phosphorylate TRAF6 and initiate downstream signaling events (Gu et al. 2013). IL-17 stimulation promotes the activation of NF-κB or mitogen-activated protein (MAP) kinases, which are essential in controlling the magnitude of immune induction (Gu et al. 2013). Several variants within the TRAF3IP2 gene locus have been associated with inflammatory diseases such as psoriasis, inflammatory bowel disease, and mucocutaneous candidiasis (Ellinghaus et al. 2010; Hüffmeier et al. 2010; Ciccacci et al. 2013; Marujo et al. 2021). A missense mutation within the TRAF3IP2 locus was associated with an inferior protection against Candida albicans (Boisson et al. 2013). The loss-of-function TRAF3IP2 variants that abolish the homeostatic activity of this IL-17R adaptor protein were associated with a compensatory hyperproduction of other T-helper cell (Th)17 cytokines, including IL-17F, IL-21, and IL-22 or dampened mucosal barrier defense (Sønder et al. 2012; Wang et al. 2013).
As the role of TRAF3IP2 in periodontal disease has not been delineated yet, the association between the combined overall variants within the TRAF3IP2 locus and the PCT3 periodontitis trait prompted us to conduct an in-depth mechanistic study on the role of TRAF3IP2 in regulating gingival barrier function. Here we report that TRAF3IP2-mediated homeostatic neutrophil recruitment is critical for maintaining host defense to avert Porphyromonas gingivalis–induced bone loss. We also show that the lead or top-ranked (smallest P value) exonic SNP rs13190932 within the TRAF3IP2 locus may serve as surrogate for a loss-of-function rs33980500 variant, leading to an impaired IL-17 signaling.
Materials and Methods
Detailed description of materials and methods is included in the Appendix.
Results
Exonic Variant rs13190932 within the TRAF3IP2 Locus Was Associated with Periodontitis
We previously reported that the overall variants across the TRAF3IP2 gene locus were significantly associated with PCT3 (Offenbacher et al. 2016; Zhang et al. 2020). When variants within the TRAF3IP2 locus were analyzed individually for association with PCT3, the lead coding nonsynonymous variant was rs13190932 (G/A), which is located in the second exon of the TRAF3IP2 gene (P = 8.43 × 10−7) (Zhang et al. 2020). The variant A allele has an overall frequency of 0.058 in the population and is more common in Caucasians, with a frequency of 0.061. Among ARIC subjects who had a complete periodontal exam and genotyping data, homozygous carriers (n = 15) of the rare allele (A/A or “2.2”) for rs13190932 had significantly more prevalent disease, defined by the percentage of interproximal sites with the attachment loss equal to or greater than 3 mm, than noncarriers (n = 4,152) (G/G or “1.1”) or heterozygous participants (n = 599) (G/A or “1.2”) (Fig. 1A).
We also found that subjects with a higher microbial burden (top 25th percentile) of the “red complex” bacteria (P. gingivalis, Tannerella forsythia, and Treponema denticola) (Fig. 1B), the “orange complex bacteria” (Campylobacter rectus, Fusobacterium nucleatum, Prevotella intermedia, and Prevotella nigrescens) (Fig. 1C), and A. actinomycetemcomitans (Fig. 1D) manifested significantly more sites with severe disease than subjects with a lower plaque pathogen burden (bottom 75th percentile) regardless of TRAF3IP2 genotypes, which was well expected. Notably, we observed that in subjects with a higher pathogen burden, the presence of 1 or 2 copies of variant A allele at rs13190932 was significantly associated with a more severe disease phenotype (Fig. 1B–D, Appendix Fig. 1). However, in subjects with a lower plaque pathogen burden, the impact of the variant was modest. These data suggest that rs13190932 (A) variant carriers are more susceptible than nonvariant allele carriers to the high plaque microbial burden–associated severe periodontitis.
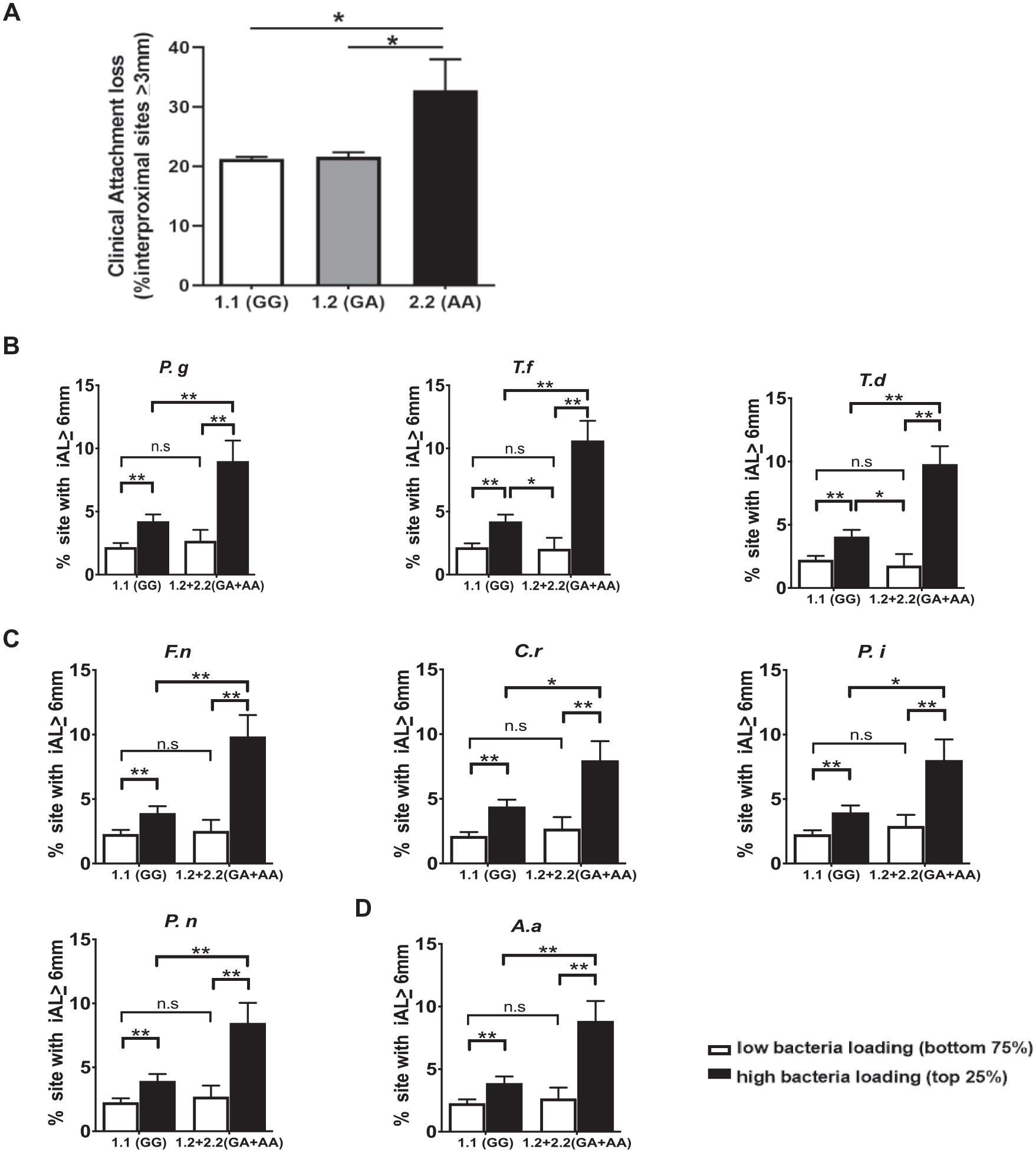
TRAF3IP2 single-nucleotide polymorphism rs13190932 is associated with periodontal disease. (
IL-17–TRAF3IP2–Mediated Response in Gingival Epithelial Cells
TRAF3IP2 and IL17R are predominantly expressed in epithelial cells in both healthy and periodontitis gingival biopsy specimens (Fig. 2A). We reported that TRAF3IP2 was indispensable for the IL-17–induced neutrophil specific chemokine CXCL1 expression in primary human gingival epithelial cells (pHGEs) (Sun et al. 2020). Here we found that an efficient knockdown of TRAF3IP2 by shRNA1 abrogated the IL-17–induced transcription of IL8 (CXCL8), CCL2, and IL6 in pHGEs (Fig. 2B). However, transcription of the matrix metalloproteinase-3 (MMP3) was not affected by the IL-17–TRAF3IP2 axis. The measurement of proteins in the supernatant confirmed the transcriptional data (Fig. 2C). TRAF3IP2 knockdown also inhibited the production of granulocyte macrophage colony-stimulating factor (GM-CSF), an important upstream signal for myeloid cell proliferation. We further identified that the activation of the canonical NF-κB pathway, as assayed by p-NF-κBp65, was involved in the IL-17–TRAF3IP2 signaling in pHGEs (Appendix Fig. 2A). Pretreatment of pHGEs with Bay 11-7082, an NF-κB inhibitor, blocked more than 50% of the CXCL1, IL8, and IL6 induction by IL-17 as compared to dimethyl sulfoxide (DMSO)–treated cells (Appendix Fig. 2B).
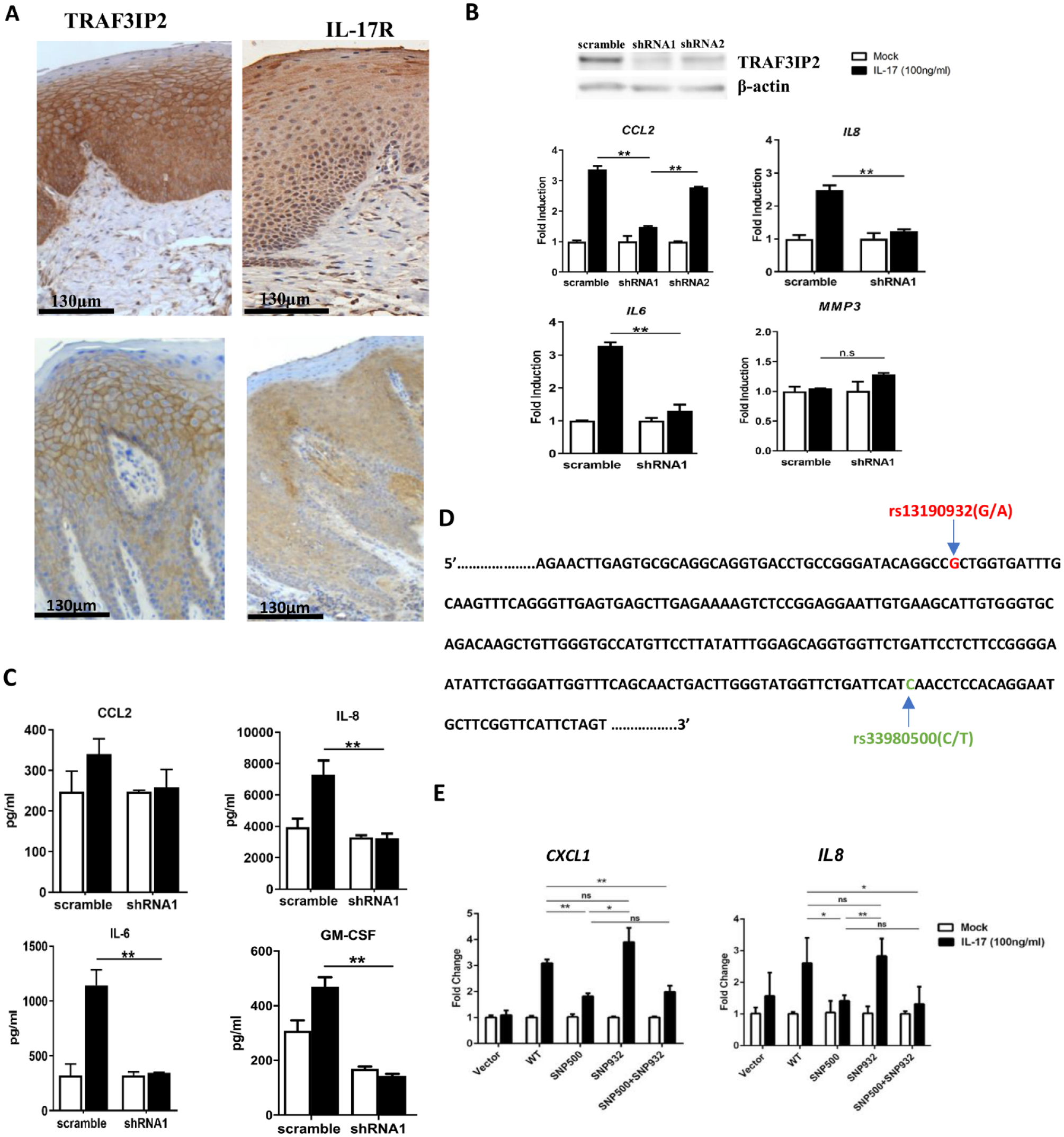
rs13190932 may serve as a proxy for the exonic rs33980500 variant within the TRAF3IP2 locus in mediating chemokine response stimulated by interleukin (IL)–17 in primary human gingival epithelial cells (pHGEs). (
The rare allele (A) frequency for rs13190932 is in high linkage disequilibrium (D′ = 1, r2 = 0.90) in North European descendants with rs33980500 (C/T), another missense nonsynonymous variant within the TRAF3IP2 locus (Fig. 2D) (Myers et al. 2020). Therefore, we assessed the functional variant effect of both SNPs on the IL-17 response. Cells transfected with the TRAF3IP2 clone bearing the variant rs13190932 (A) stimulated by IL-17 resembled the cells transfected with the nonvariant TRAF3IP2 clone, while the variant rs33980500 (T) significantly blocked the transcription of CXCL1 and IL8 in comparison to the nonvariant transfected cells (Fig. 2E). The cellular response to the variant haplotype with both variants was similar to the response of the single rs33980500 variant. These data suggest that, rather than rs13190932, rs33980500 is a loss-of-function variant and might be a genuine periodontal disease–associating variant.
Traf3ip2–/– Mice Were More Susceptible to P. gingivalis–Induced Periodontitis That Is Associated with a Compromised Mucosal Defense Transcriptomic Signature
We next employed a pathogen-induced oral infection model to characterize the role of the homeostatic TRAF3IP2-mediated IL-17 pathway in periodontitis (Fig. 3A). Traf3ip2–/– mice that were orally challenged with the P. gingivalis A7436 strain presented significantly more alveolar bone loss than Traf3ip2–/– mice challenged with Carboxymethylcellulose CMC (control) (Fig. 3B). However, no significant difference was found between P. gingivalis A7436 and CMC-challenged WT mice. The resistance to P. gingivalis–induced bone loss in the C57BL/6 background WT mice was consistent with previous reports (Baker et al. 2000; Sun et al. 2020). The average net bone loss as the bone level subtracting the CMC challenged from the P. gingivalis–challenged Traf3ip2–/– mice was also significantly higher than the net bone loss calculated from WT mice (Fig. 3B). This net bone loss reflects the strain-specific susceptibility to P. gingivalis–induced bone loss.
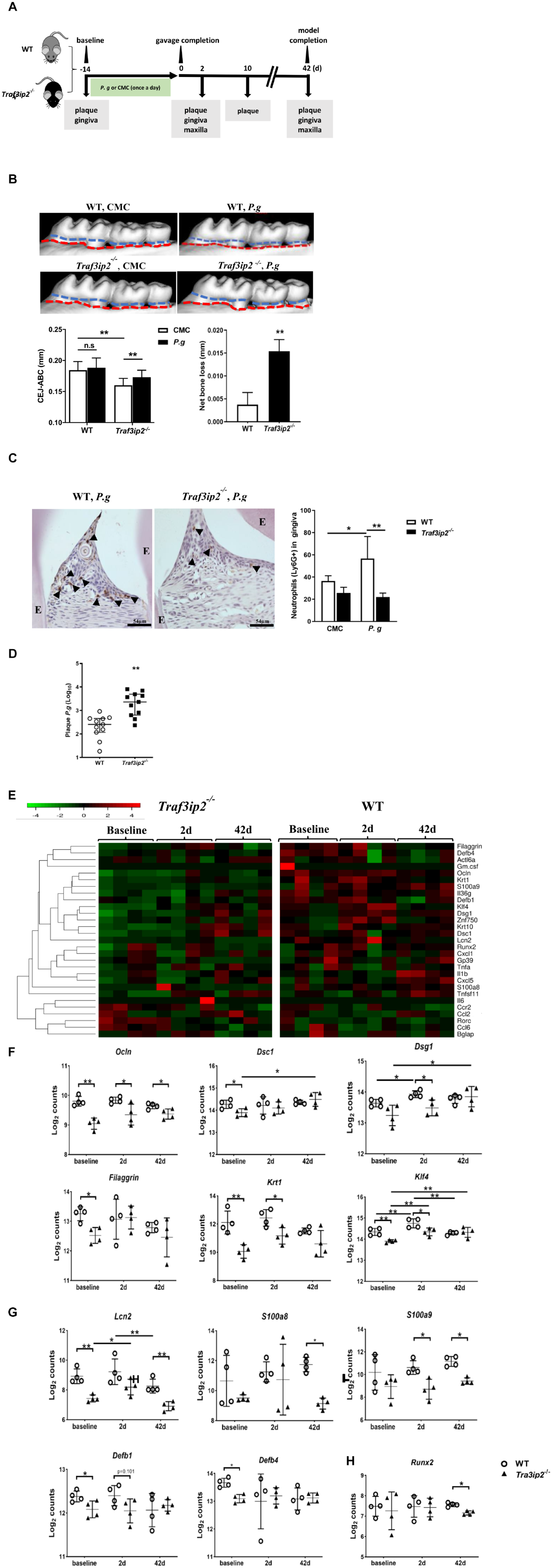
Traf3ip2–/– mice were more susceptible to Porphyromonas gingivalis–induced periodontitis than wild-type (WT) mice. (
We next compared the neutrophil recruitment in Traf3ip2–/– mouse gingiva to the recruitment in WT mice. Although P. gingivalis challenge resulted in a significant increase in neutrophils as compared to CMC treatment in WT mice, significantly fewer Ly6G+ neutrophils were present in the gingival tissue from Traf3ip2–/– mice than WT mice 2 d after the completion of the P. gingivalis challenge (Fig. 3C). In addition, the residual plaque P. gingivalis was significantly higher in Traf3ip2–/– mice than WT mice at the same time point (Fig. 3D).
However, in a ligature-induced periodontitis mouse model, the bone level was not significantly different between WT and Traf3ip2 null mice (Appendix Fig. 3). Taken together, our in vivo data suggest that the loss of the homeostatic IL-17–TRAF3IP2 pathway predisposes mice to the P. gingivalis–induced alveolar periodontal bone loss, impaired neutrophil recruitment in gingiva, and delayed the clearance of P. gingivalis.
We also assessed the IL-17 pathway-associated transcriptomic profile in Traf3ip2–/– mouse gingiva at different stages upon P. gingivalis challenge (Fig. 3E). We did not observe significant changes in transcripts of chemokine or cytokine genes, including Cxcl1, Ccl2, Gmcsf, and Il6, in the Traf3ip2–/– mouse gingiva as compared to the WT control animals. Instead, we found that the transcription of several epithelial marker genes and those associated with epithelial barrier function, including keratin 1 (Krt1), Kruppel-like factor 4 (Klf4), occludin (Ocln), desmocollin 1 (Dsc1), desmoglein 1 (Dsg1), and Filaggrin, was significantly lower in Traf3ip2 null mouse gingiva than WT mice either at the baseline or upon P. gingivalis challenge (Fig. 3F). Transcription of genes closely associated with antimicrobial defense, including lipocalin 2 (Lcn2), S100a8, S100a9, and β defensins (Defb1 and Defb4), was also significantly diminished in the knockout (KO) mouse gingiva (Fig. 3G). The transcription of Runx2, an osteoblast differentiation marker, was significantly lower in Traf3ip2–/– mice upon P. gingivalis challenge than WT mouse gingiva. Such a transcriptomic signature in the KO animals suggested a breach in both a physical and an immune barrier in the gingiva of Traf3ip2–/– mice that were susceptible to P. gingivalis–induced alveolar bone loss.
Absence of IL-17–TRAF3IP2 Pathway Was Associated with a Distinct Oral Microbial Profile
Through the 16S ribosomal RNA (rRNA) sequencing, we found that the oral plaque from Traf3ip2–/– mice had a lower compositional (α) diversity at the baseline and during P. gingivalis oral challenge than the WT plaque samples (Appendix Fig. 4). The β-diversity from all oral plaque samples clearly illustrated a unique microbial signature in Traf3ip2–/– oral plaque (Fig. 4A). We observed that the relative abundance of taxonomic groups of bacteria was drastically different between the KO and WT mice (Fig. 4B). For example, Streptococcus was more enriched in oral plaque from Traf3ip2–/– mice at any given time point than the WT mice samples. Further Linear discriminant analysis Effect Size LEfSe analysis indicated that bacteria from the Corynebacterium and Staphylococcus genera were significantly more abundant in WT plaque, while the Streptococcus genus was significantly more enriched in Traf3ip2–/– mice prior to or upon P. gingivalis challenge (Fig. 4C, D).
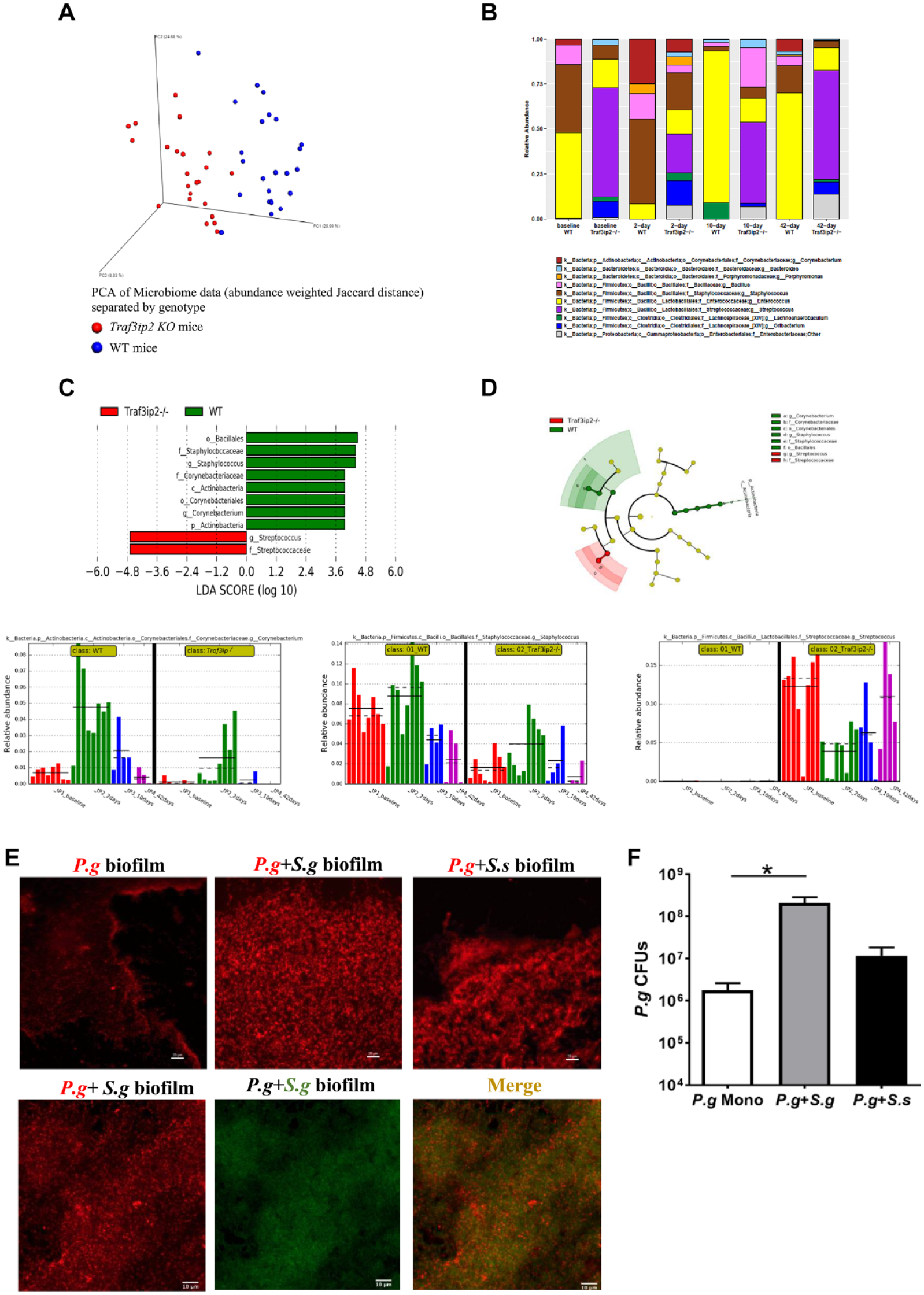
The oral plaque of Traf3ip2–/– mice harbored a unique microbial community structure. (
The high abundance of Streptococcus genus bacteria, observed in Traf3ip2–/–, may help P. gingivalis to colonize in plaque (Simionato et al. 2006). Through a fluorescence in situ hybridization (FISH) assay, we found that P. gingivalis more abundantly presented in biofilm in the presence of Streptococcus gordonii or Streptococcus sanguinis than P. gingivalis monospecies biofilm in vitro (Fig. 4E). Significantly more P. gingivalis colonization in the dual species than mono P. gingivalis biofilm was further confirmed through quantitative PCR (Fig. 4F).
Homeostatic IL-17-TRAF3IP2 Axis Strengthens Gingival Defense against Invading Oral Pathogens by Neutrophil Recruitment
We next performed a ligature-enhanced P. gingivalis local invasion experiment (Chipashvili et al. 2021). We found that gingiva from Traf3ip2–/– mice harbored significantly more total bacteria than WT mice prior to P. gingivalis challenge (Fig. 5A). We further detected a trend of more bacterial colonies in Traf3ip2–/– mouse gingival homogenate than WT mouse under an aerobic culture condition (Fig. 5B, Appendix Fig. 5). In P. gingivalis–challenged mice, the cultivatable bacterial colonies were significantly more in Traf3ip2–/– mouse gingival homogenate than WT mice under both an aerobic and anaerobic condition (Fig. 5C). P. gingivalis DNA was present in 3 out of 10 P. gingivalis–challenged WT mouse gingival tissues in the invasion model, while 7 out of 10 Traf3ip2–/– mouse gingivae were clearly positive for P. gingivalis (Fig. 5D). More P. gingivalis invasion into Traf3ip2–/– gingiva was associated with significantly more bone loss in the distal root of the first molar than that of WT mice, although the bone loss in the mesial root of the second molar was not significantly different (Fig. 5E).
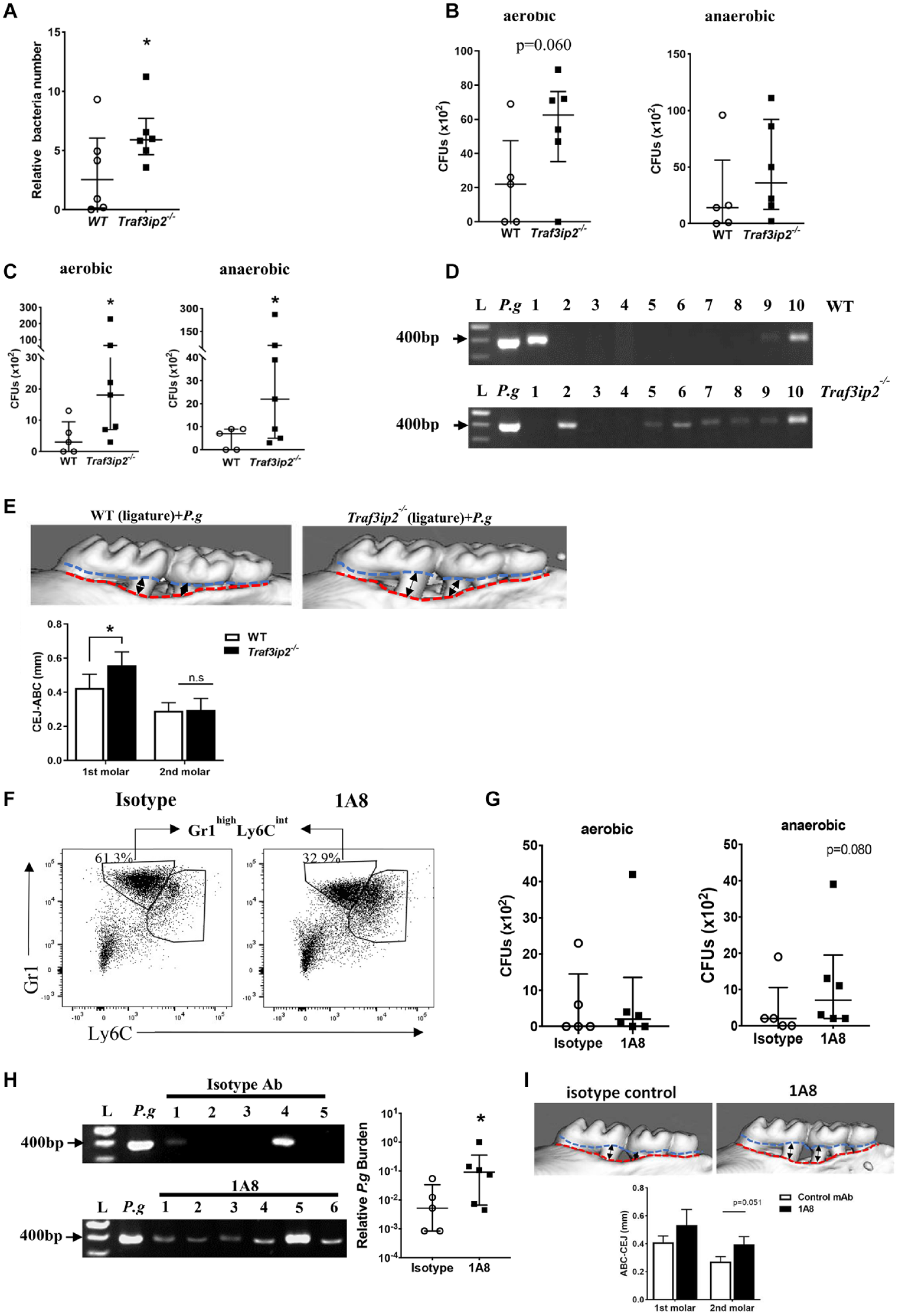
More bacteria invaded into the gingiva of Traf3ip2–/– mice than wild-type (WT) mice in the ligature-enhanced Porphyromonas gingivalis invasion model. After ligature placement, approximately 0.5 × 109 P. gingivalis or CMC carrier was applied to the mouse oral cavity. Such an oral challenge by P.g was repeated daily for 14 consecutive days. (
Based on the data in Figure 3C and G, we hypothesized that a dampened neutrophil response promoted P. gingivalis gingival invasion, leading to more severe bone loss observed in Traf3ip2–/– mice. To test this hypothesis, we depleted neutrophils in WT mice with the antibody 1A8 in the invasion model. The deletion efficiency was about 50% in gingiva and 65% in blood (Fig. 5F and Appendix Fig. 6A). We observed a trend of more cultivatable anaerobes in the gingival homogenate from the 1A8-treated mice than the isotype control antibody-treated animals after P. gingivalis challenge (Fig. 5G and Appendix Fig. 6B). While P. gingivalis DNA in the isotype antibody-treated mice was rarely detected, P. gingivalis DNA was found in all 6 gingival tissues collected from the neutrophil-depleted mice (Fig. 5H, left panel). Significantly more P. gingivalis invaded into the gingiva in neutrophil-depleted mice through quantitative polymerase chain reaction (qPCR) quantification (Fig. 5H, right panel). The 1A8-treated mice exhibited more alveolar bone loss than the isotype control antibody-treated mice (Fig. 5I). Overall, these data strongly suggest that the reduced neutrophil recruitment due to the absence of the TRAF3IP2-mediated IL-17 pathway in mouse gingiva permitted more P. gingivalis to invade into gingiva that resulted in more bone resorption.
Discussion
Recent studies frequently implicated an excess of IL-17 signaling in the pathogenesis of periodontitis. However, using Traf3ip2–/– mice simulating the extremity of a loss-of-function variant effect, we demonstrate that an integral IL-17–TRAF3IP2 signaling axis protects the host against the pathogen-induced periodontitis. Yu et al. (2007) reported that an absence of the IL-17 pathway in Il17r(a)–/– mice increased the animal’s susceptibility to P. gingivalis–induced periodontitis. Therefore, fine-tuning the TRAF3IP2-mediated IL-17 response is fundamental to avoid the inflammatory insult while enhancing mucosal immune defense against invading periodontal pathogens.
Neutrophils patrolling at the mucosal surface play a pivotal role in defending mucosal barriers by eliminating invading pathogens (Conti et al. 2009; Hajishengallis et al. 2015; Uriarte et al. 2016). A reduced neutrophil recruitment to the gingiva in Traf3ip2–/– mice was associated with a delayed clearance of P. gingivalis and an increased local tissue invasion by this pathogen. The increased P. gingivalis inside the Traf3ip2–/– mouse gingiva was also possibly due to the pathogen retention in the unresolved periodontitis lesion. P. gingivalis A7436 is a virulent strain excelling at evasion of phagocytosis. This strain of P. gingivalis was shown to be notoriously persistent in cells upon invasion and a weak inducer of inflammation (Genco et al. 1991; Rodrigues et al. 2012). Our transcriptomic data also indicated that the P. gingivalis A7436 did not induce a more inflammatory response in Traf3ip2–/– than WT mice (Fig. 3E). However, a reduced neutrophil response in the Traf3ip2 KO mice further aggravated the invasion by P. gingivalis A7436, a strain that is capable of “sneaking through” the mucosal defense barrier.
In this report, we used 3 murine models to assess the role of the TRAF3IP2–IL-17 axis in periodontitis. The widely used classical P. gingivalis oral challenge model, in which bone loss is usually moderate and slow progressing, mimics the overall host-specific pathogen interactions in human periodontitis. We found that a lack of neutrophil response was associated with more bone loss in Traf3ip2–/– mice upon P. gingivalis challenge. In the ligature-induced periodontitis model, which is fast-progressing and manifests more inflammation than the oral challenge model, the ligature promotes the aggregation of nonspecific plaque bacteria and amplifies gingival inflammatory responses. This model is typically used to evaluate the host inflammatory response. However, the bone loss of Traf3ip2–/– mice in this model was not more severe than the WT mice (Appendix Fig. 3). A similar bone destruction in the Traf3ip2–/– mice to the WT mice suggests that the lack of IL-17 inflammatory response may be compensated by other inflammatory pathways such as the Toll-like receptor (TLR)–mediated immune response. Other immune cells than neutrophils, such as macrophages, also play a critical role in the bone loss in the ligature model (Sun et al. 2020). In the ligature-enhanced P. gingivalis invasion model, the ulceration of the sulcus epithelium induced by ligature facilitates more P. gingivalis intra-tissue invasion than the oral challenge alone (Graves et al. 2008). Combined with neutrophil depletion, this model allows us to study the specific role of neutrophils in P. gingivalis invasion. The inoculation of P. gingivalis in the invasion model promoted more anaerobic bacteria invasion and aggravated the bone loss in Traf3ip2–/– mice in comparison to WT mice. These data indicate that the local gingiva with a weak neutrophil-mediated barrier in Traf3ip2–/– mice is particular susceptible to the pathogenic bacteria such as P. gingivalis–induced bone loss. We also noted that the steady state of alveolar bone loss (CEJ–ABC) in Traf3ip2–/– mice was less than that of the WT mice (Fig. 3B). This is likely due to the fact that IL-17 is also an upstream bone resorption signal. It was shown that the loss of TRAF3IP2 had a protective effect for bone resorption in a non-infection post menopausal osteoporosis model (DeSelm et al. 2012).
Several GWA studies have identified novel candidate periodontitis-associating variants and genetic loci, most of which have not been previously implicated in periodontal disease (Divaris et al. 2013; Munz et al. 2017; Tong et al. 2019). Although neither the rs13190932 variant nor the rs33980500 variant was previously associated with periodontal disease, both variants have been repeatedly reported to be risk alleles for psoriasis and psoriatic arthritis (PA) in certain populations (Ellinghaus et al. 2010). Therefore, the shared variants identified by both periodontitis and psoriasis may suggest that the TRAF3IP2-mediated IL-17 pathway critically participates in the inflammatory response and barrier defense at mucocutaneous surfaces. Several clinical reports demonstrated that psoriasis or PA patients had more severe periodontal disease than subjects without psoriasis or PA (Skudutyte-Rysstad et al. 2014; Zhang et al. 2022). Therefore, the shared risk of periodontal disease and psoriasis modified by TRAF3IP2 variants suggests a common etiopathogenic mechanism involving the IL-17–TRAF3IP2 axis that likely links oral and systemic diseases. Our data also suggested that a severe bone loss upon P. gingivalis challenge was associated with a compromised epithelial barrier. We found decreased transcription of several epithelial barrier and homeostatic marker genes in the Traf3ip2–/– gingiva without P. gingivalis challenge, indicating a potentially compromised epithelial barrier at the baseline level. Through RNA sequencing, Lambert et al. (2017) noted that the expression of several epithelial differentiation genes, including those that we reported here, was also downregulated in TRAF3IP2-silenced keratinocytes. Our data warrant further studies investigating the role of TRAF3IP2-mediated gingival epithelial barrier function in periodontitis.
We identified a less diverse microbial community structure in the oral plaque of Traf3ip2–/– mice than WT mice, which was associated with a decreased transcription of several antimicrobial genes in those mice. This finding contrasts several subgingival microbiome analyses in human subjects, which usually show a more diverse microbial community in periodontitis patients than periodontal health (Griffen et al. 2012; Camelo-Castillo et al. 2015). The dysbiotic pattern in P. gingivalis–induced periodontitis in a murine model may not be readily translatable to human periodontitis. A less diverse microbiome structure may be also inherently characteristic of a compromised gingival barrier in a reduced IL-17–TRAF3IP2–neutrophil response. Several studies have reported that mixed oral infection by S. gordonii and P. gingivalis in mice resulted in a more significant alveolar bone loss than P. gingivalis monoinfection (Daep et al. 2011; Kuboniwa et al. 2017). The more abundant Streptococcus bacteria in Traf3ip2–/– mouse oral plaque may drastically enhance the plaque biofilm colonization by P. gingivalis (Maeda et al. 2008; Daep et al. 2011).
We acknowledge that the findings of this study might be limited to the A7436 strain of P. gingivalis. Other commonly used strains need to be tested to confirm whether the homeostatic IL-17–TRAF3IP2–mediated immune response is a common mechanism to enhance mucosal defense. However, based on the similar biological structure and behavior of A7436 to other strains such as W83, we would expect that the disease mechanism identified using the A7436 strain in our models can be generalizable to other P. gingivalis strains (Chastain-Gross et al. 2015).
In summary, we demonstrated that the lead exonic variant rs13190932, which is likely a surrogate SNP for a genuine loss-of-function variant rs33980500, within the TRAF3IP2 locus was significantly associated with more severe periodontitis in carriers with a high microbial pathogen burden than noncarriers. Animal model data further delineated an essential role of the IL-17–TRAF3IP2–neutrophil axis in safeguarding the gingival immune barrier against P. gingivalis invasion and therefore mitigating the P. gingivalis–induced alveolar bone loss. In addition, the loss of TRAF3IP2 in mice was characteristically associated with a distinct oral microbial community structure.
Author Contributions
J. Zhang, L. Sun, contributed to design, data analysis, drafted and critically revised the manuscript; M.H.H. Withanage, contributed to data analysis, drafted and critically revised the manuscript; S.M. Ganesan, F.R. Teles, contributed to data analysis and interpretation, critically revised the manuscript; M.A. Williamson, contributed to data acquisition and analysis, critically revised the manuscript; J.T. Marchesan, contributed to conception, data analysis, critically revised the manuscript; Y. Jiao, contributed to conception, data acquisition, critically revised the manuscript; N. Yu, contributed to data acquisition and interpretation, critically revised the manuscript; Y. Liu, D. Wu, K.L. Moss, contributed to data analysis, critically revised the manuscript; A.K. Mangalam, Y.L. Lei, contributed to data conception, critically revised the manuscript; E. Zeng, contributed to data analysis and interpretation, drafted the manuscript; S. Zhang, contributed to conception and design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345221123256 – Supplemental material for TRAF3IP2–IL-17 Axis Strengthens the Gingival Defense against Pathogens
Supplemental material, sj-docx-1-jdr-10.1177_00220345221123256 for TRAF3IP2–IL-17 Axis Strengthens the Gingival Defense against Pathogens by J. Zhang, L. Sun, M.H.H. Withanage, S.M. Ganesan, M.A. Williamson, J.T. Marchesan, Y. Jiao, F.R. Teles, N. Yu, Y. Liu, D. Wu, K.L. Moss, A.K. Mangalam, E. Zeng, Y.L. Lei and S. Zhang in Journal of Dental Research
Footnotes
Acknowledgements
The Atherosclerosis Risk in Communities (ARIC) Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HSN268201100012C), R01HL087641, R01HL59367, and R01HL086694; National Human Genome Research Institute contract U01HG004402; National Institutes of Health (NIH) contract HHSN268200625226C; National Institute of Environmental Health Sciences grant P30ES010126; and National Institute of Dental and Craniofacial Research grants R01DE11551 and R01DE021418. We deeply thank the staff and participants of the ARIC study for their contributions. We thank Dr. Ulrich Siebenlist at the National Institute of Allergy and Infectious Diseases for providing Traf3ip2(Act1, CISK)–/– and wild-type control animals. We also thank the Center for Gastrointestinal Biology and Disease (CGIBD P30 DK034987) and the UNC Nutrition Obesity Research Center (NORC P30 DK056350). We want to recognize Dr. Steven Offenbacher, a pioneer in periodontal research whose leadership and expertise contributed to the early conceptualization of this research. The data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center.
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by NIH grants R00 DE027086 (to S.Z.), K01 DE027087 (to J.T.M.), R01 DE026728 and U01 DE029255 (to Y.L.L.), and F32 DE026688 (Y.J.).
Data Availability
All data published in this article are available upon reasonable request.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.