
Abstract

Background
Congenital chloride diarrhea (CCD, OMIM 214700) is a rare autosomal recessive disorder caused by loss-of-function mutations in the SLC26A3 gene (SLC26A3; OMIM *126650), which encodes a protein that exchanges chloride and bicarbonate on enterocyte apical surface.1,2 Patients usually have a history of antenatal polyhydramnios and may experience dilated intestinal loops, watery stool, dehydration, hypochloremic hypokalemic metabolic alkalosis, and high fecal chloride excretion. 3 The presence of abdominal distention, absence of meconium, and enlarged intestinal loops may lead to misdiagnosis of meconium ileus or Hirschsprung’s disease, resulting in unnecessary surgical procedures. 4 However, patients with CCD may also be misdiagnosed as Bartter and Pseudo Bartter syndrome due to the similar laboratory findings.
Educational Objectives
Due to similar clinical and laboratory presentations, congenital chloride diarrhea (CCD) and Hirschsprung’s disease may be misdiagnosed as each other.
Before diagnosing a disease that may cause intestinal obstruction and require urgent surgery, such as Hirschsprung’s disease, it is necessary to keep in mind other potential diagnoses, such as CCD, that can be managed with medical treatment options without the need for surgery.
Case Presentation
A female patient with dilated intestinal loops and polyhydramnios on antenatal ultrasonography (US) was born at the 36th gestational week from healthy and unrelated parents with a birth weight of 2350 g. She was admitted to the neonatal intensive care unit because of severe abdominal distention and respiratory problems. The patient did not have meconium passage in the first 24 hours. Ileal atresia was excluded by unprepped barium enema X-ray, but revealed a suspicious transition zone, and she was operated on the postnatal 20th day with the diagnosis of Hirschsprung’s disease. However, the pathology samples revealed that all intestinal segments had ganglion cells after surgery. No persistent constipation or diarrhea was observed after the surgery, and the patient was discharged with a weight of 2600 g at 3 months of age. She had no diarrhea between the third and fourth months. She experienced intermittent diarrhea from the fourth to the sixth month after receiving iron prophylaxis and rotavirus vaccination in the fourth month. At 6 months of age, the patient was admitted to a local hospital with complaints of persistent diarrhea, lethargy, and weight loss, where she was noted to have hypochloremic hypokalemic metabolic alkalosis and hyponatremia. She was diagnosed with Bartter syndrome and referred to our clinic.
At the time of admission, she had severe watery diarrhea, mild dehydration, and severe malnutrition (weight: 4700 g, < −3 standard deviation; height 63 cm, −1.22 standard deviation). Venous blood gas, serum, urine, stool electrolytes analyses, renin, and aldosterone levels were studied. The results of the patient’s initial laboratory examinations are shown in Table 1.
The Results of the Patient’s Initial Laboratory Examinations.
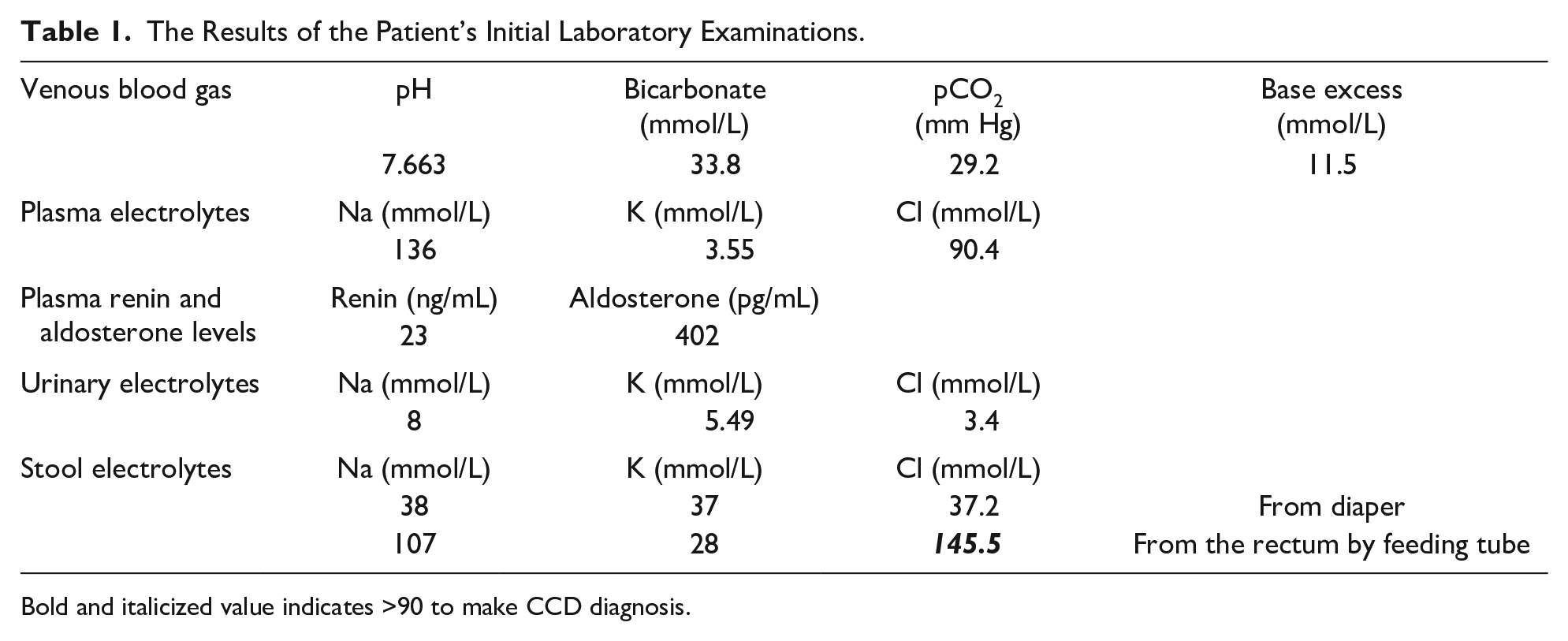
Bold and italicized value indicates >90 to make CCD diagnosis.
Bartter syndrome was excluded with inappropriate plasma renin, aldosterone levels, and low urinary electrolytes; cystic fibrosis was excluded with a normal sweat chloride level and negative genetic testing. The absence of conditions that could lead to Pseudo Bartter syndrome, such as diuretic drug use, long-term chlorine-poor diet, cyclic vomiting attacks, and long-term laxative drug use, and the exclusion of cystic fibrosis led us to consider the diagnosis of CCD. We double-checked for CCD (although the results are shown normally in the first stool sample taken from the diaper), and a second examination of the liquid stool sample was obtained by entering the rectum with a feeding tube. In this sample, stool chloride was 145.5 mmol/L, stool sodium was 107 mmol/L, stool potassium was 28 mmol/L, and stool osmolar gap was 20, supporting the diagnosis of CCD. In our patient, a heterozygous mutation detected between nucleotides 2024 and 2026 in exon 18 (c.2024_2026dup TCA) of the SLC26A3 gene that was characterized by duplication of 3 nucleotides (thymidine, cytosine, and adenine). This pathogenic mutation was also present in our patient’s mother. However, a rare heterozygous mutation in the SLC26A3 (c.1954G>A), which was characterized by the replacement of a guanine by an adenine at nucleotide 1954 in exon 17, was also detected in our patient and her father.
She was primarily treated with intravenous fluids and then started on oral sodium chloride (5 mmol/kg/day), potassium chloride (2 mmol/kg/day), and lansoprazole (1 mg/kg/day). Her condition improved, and the frequency of diarrhea decreased. The patient is about to turn 3 years old, has an average weight, and takes oral supplements of sodium chloride and potassium. She regularly attends outpatient clinic appointments.
Discussion
The primary defect in CCD is the disruption of the Cl−/HCO3− exchange on the duodenal, ileal, and colonic epithelial brush border caused by mutations in the SLC26A3 gene. 5 Active reabsorption of chloride from the intestines is defective, and there is an increased excretion of chloride in the stool, resulting in hypochloremia. Inadequate bicarbonate secretion inhibits sodium absorption via the Na/H exchanger, leading to metabolic alkalosis and acidification of the intestinal contents. Secondary hyperaldosteronism develops due to sodium and water loss, producing potassium excretion and further exacerbating hypokalemia and hyponatremia. 6 The diagnosis of our patient was confirmed by hypochloremic hypokalemic metabolic alkalosis, hyponatremia, and higher fecal chloride content. Our patients’ fecal chloride level was above 90 mmol/L, which is necessary for diagnosis.
This congenital metabolic disease is more common in countries with higher consanguinity rates. More than 70 gene mutations, including mutations originating from countries with a high prevalence of CCD, are noted in the human gene mutation database (http://www.hgmd.cf.ac.uk). Many of these mutations are single nucleotide variations or small deletions, many of which affect exons 3-6 and exons 12-15. 7 In our patient, 2 mutations in the compound heterozygous state were detected at the SLC26A3 gene. One of these mutations was pathogenic c.2024_2026dupTCA (p.(Ile675dup)), which is also characteristic of half of Polish CCD patients. 8 This classic mutation was reported in other countries as well. Besides that, a rare missense c.1954G>A (p.(Asp652Asn)) variant that was classified as likely pathogenic was identified in the heterozygous state of our patient. This rare mutation has been reported in 1 Turkish homozygous case 9 and detected in 4 other cases worldwide. 2 Segregation analysis revealed that the c.2024_2026dupTCA (p.(Ile675dup)) variant was maternally, and the c.1954G>A (p) variant was paternally inherited. This is the first Turkish CCD case with compound heterozygous mutations in the SLC26A3 gene.
In CCD, watery diarrhea starts in fetal life. The affected fetus may be diagnosed by antenatal ultrasound revealing polyhydramnios and dilated, fluid-filled, intestinal loops. 10 Some patients with CCD may have low birth weight or be born prematurely. Abdominal distension and the absence of shaped meconium are vital signs that can lead to the diagnosis. The lack of particles in stool and its close resemblance to urine could be mistaken for the absence of meconium.11-13 Similarly, our patient had a false diagnosis of Hirschsprung’s disease. Watery diarrhea must be evaluated by taking it from the rectum with a feeding tube. Surgical treatment should not be considered before a definite diagnosis of Hirschsprung’s disease verified by a transition zone on unprepped barium enema X-ray, abnormal internal anal sphincter resting pressure and rectoanal inhibitory reflex on anorectal manometry, and absence of ganglion cells on rectal biopsy. 14 Unfortunately, our patient was considered to have Hirschsprung’s disease and required surgery due to the suspicious transition zone detected on an unprepped barium enema X-ray, which was consistent with the clinical findings. It was learned that anorectal manometry could not be performed because she was too young and could not adapt.
Detection of dilated intestinal loops and polyhydramnios in the prenatal US during the third trimester of pregnancy are the most prominent signs of intestinal obstruction, especially jejunal and ileal atresia. Other clinical manifestations such as meconium ileus, colonic atresia, Hirschsprung’s disease, and imperforate anus may also occur with similar ultrasound features.15,16 In recent years, Colombani et al 17 reported that fetal magnetic resonance imaging (MRI) helps reveal the diagnosis in addition to ultrasound findings. They stated that in CCD patients, it is an important finding that fluid-filled intestines are observed as hypointense due to watery diarrhea instead of meconium, which usually appears hyperintense on T1-weighted images on MRI. In 2017, Kawamura and Nishiguchi presented a CCD case that showed fetal intestinal dilatation findings (honeycomb sign) in ultrasonography at the 26th gestational week. Their patient had a large amount of hypointense fluid throughout the rectum and intestinal dilatation in MRI. 18 Ultrasonography detected polyhydramniosis and intestinal dilatation in our patient. Fetal MR imaging may aid in differentiating CCD as a potential diagnosis.
Congenital chloride diarrhea can easily be misdiagnosed as Bartter syndrome due to similar laboratory findings. However, the primary defect in Bartter syndrome is in the triple electrolyte cotransporter (Na/K/2Cl) in the thick ascending arm of Henle. Bartter syndrome is characterized by polydipsia, polyuria, increased excretion of sodium and chloride in urine, and dehydration, accompanied by vomiting and constipation. Laboratory tests show hypochloremic hypokalemic metabolic alkalosis and increased aldosterone and renin levels.19,20 Our patient was referred to us after being misdiagnosed with Bartter syndrome 5 days prior. At our hospital, we excluded Bartter syndrome through normal urinary electrolytes and serum aldosterone levels. A slightly elevated renin level was attributed to mild dehydration in the patient. When Bartter syndrome is ruled out, Pseudo Bartter syndrome may be considered due to similar clinical presentation. It differs from Bartter syndrome because biochemical changes occur without tubular dysfunction in Pseudo Bartter syndrome. The leading cause of Pseudo Bartter syndrome is cystic fibrosis. Still, it can also result from CCD, Gitelman syndrome, diuretic drug use, long-term chlorine-poor diet, cyclic vomiting attacks, and long-term laxative drug use. 21 In our patient, CCD was diagnosed and confirmed by examinations after excluding cystic fibrosis and other etiological causes of Pseudo Bartter syndrome.
Patients with CCD may experience chronic diarrhea, failure to thrive, acute dehydration attacks in infections, psychomotor delay, chronic kidney disease, male subfertility, and inflammatory bowel disease. The most critical points to prevent the development of all these negative prognostic pictures are regular NaCl/KCl supplementation, periodic evaluation of serum electrolytes, renin and aldosterone, growth parameters, renal functions, and psychomotor development, vaccination compliance, and intermittent examination for infertility. 1
Conclusion
Congenital chloride diarrhea is a rare disease that is included in the differential diagnosis of hypochloremic metabolic alkalosis in childhood. Congenital chloride diarrhea should also be kept in mind in patients with dilated intestinal loops on the prenatal US and delayed meconium passage to avoid unnecessary surgery.
Author Contributions
ATÇ and ZB: provided clinical care to the patient, performed literature review, drafted the initial manuscript; and reviewed and revised the manuscript. YA and SK: performed literature review, critically reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of work.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Written informed consent for patient information to be published was provided by a legally authorized representative.