
Abstract
Gaucher disease (GD) is a rare autosomal recessive metabolic disorder. It is characterized by a deficiency of lysosomal glucocerebrosidase, which results in the accumulation of glycosphingolipid substrates, primarily glucosylceramide, in the phagocyte system. In GD Type 1, the liver, spleen, and bone marrow are typically affected. We report the case of a 7-year-old female with GD Type 1 who presented with hepatosplenomegaly detected incidentally following a motor vehicle accident. She was found to have concomitant thrombocytopenia and Erlenmeyer flask deformities of her lower limbs. Diagnosis was made on the basis of very low leukocyte β-glucocerebrosidase activity and elevated plasma chitotriosidase. DNA mutation studies revealed both c.1226A>G and c.116_1505 deletion (exons 3-11). The patient is currently managed with biweekly intravenous imiglucerase (Cerezyme) replacement therapy. She demonstrated resolution of thrombocytopenia and hepatosplenomegaly at 2-year follow-up. Physicians must consider this rare diagnosis in children presenting with hepatosplenomegaly to prompt timely management.
Introduction
Gaucher disease (GD) is the most common lysosomal storage disorder. 1 It is autosomal recessively inherited and results from pathological variants in the GBA gene, which codes for the lysosomal enzyme β-glucocerebrosidase (acid β-glucosidase). Deficiency of this enzyme results in accumulation of its major substrate, glucocerebroside, within the lysosomes of macrophages. These pathologic macrophages, known as Gaucher cells, infiltrate the liver, spleen, bone marrow, lungs, and occasionally nervous system. 1
There are 3 main types of GD, including nonneuronopathic (Type 1), acute neuronopathic (Type 2), and chronic neuronopathic (Type 3). Type 1 is the most prevalent and accounts for up to 90% of all cases. 2 It is characterized by multisystem involvement including hepatosplenomegaly, bony pain, osteonecrosis, pathological fractures, anemia, thrombocytopenia, fatigue, and failure to thrive. 2
Enzyme replacement therapy (ERT) with intravenous recombinant mannose-terminated glucocerebrosidase is first-line treatment for GD Type 1. Due to its reversible effects on visceral damage, ERT has the potential to reduce hepatosplenomegaly, minimize skeletal pathology, correct anemia and thrombocytopenia, and optimize growth. 3 In this report, we describe a rare case of incidentally detected GD Type 1 treated with imiglucerase replacement therapy.
Case Presentation
A 7-year-old female was referred to the Paediatric Liver Clinic for hepatosplenomegaly found incidentally on assessment following a minor motor vehicle accident. The patient was asymptomatic aside from endorsing a history of easy bruising, pain to her upper and lower limbs, and occasional epistaxis. Developmentally, she was age appropriate with no neurological symptoms, fevers, weight loss, night sweats, or manifestations of chronic liver disease. Her past medical history was significant only for asthma and recurrent tonsillitis. The patient’s parents were nonconsanguineous. Her mother was of Scottish, Irish, and possibly Turkish descent, and her father was Ethiopian. Family history was significant for amyotrophic lateral sclerosis in the biological father.
On physical examination, the patient’s vital signs were stable. Her weight was 23.7 kg (50th percentile) and height was 118.3 cm (15th percentile). She did not have any dysmorphic features. Her abdomen was soft and nontender with a liver palpable 4 cm below the right costal margin. Her splenic edge was palpable at the umbilicus. There was no evidence of anemia, jaundice, finger clubbing, or lymphadenopathy. Neurological examination was unremarkable.
An ultrasound of her abdomen demonstrated an elongated spleen of 16 cm and liver of 14 cm. Parenchymal echogenicity of the liver and spleen were satisfactory. Color Doppler studies did not detect any hemodynamic lesions. As seen in Figure 1, the differential diagnosis for hepatosplenomegaly is broad.
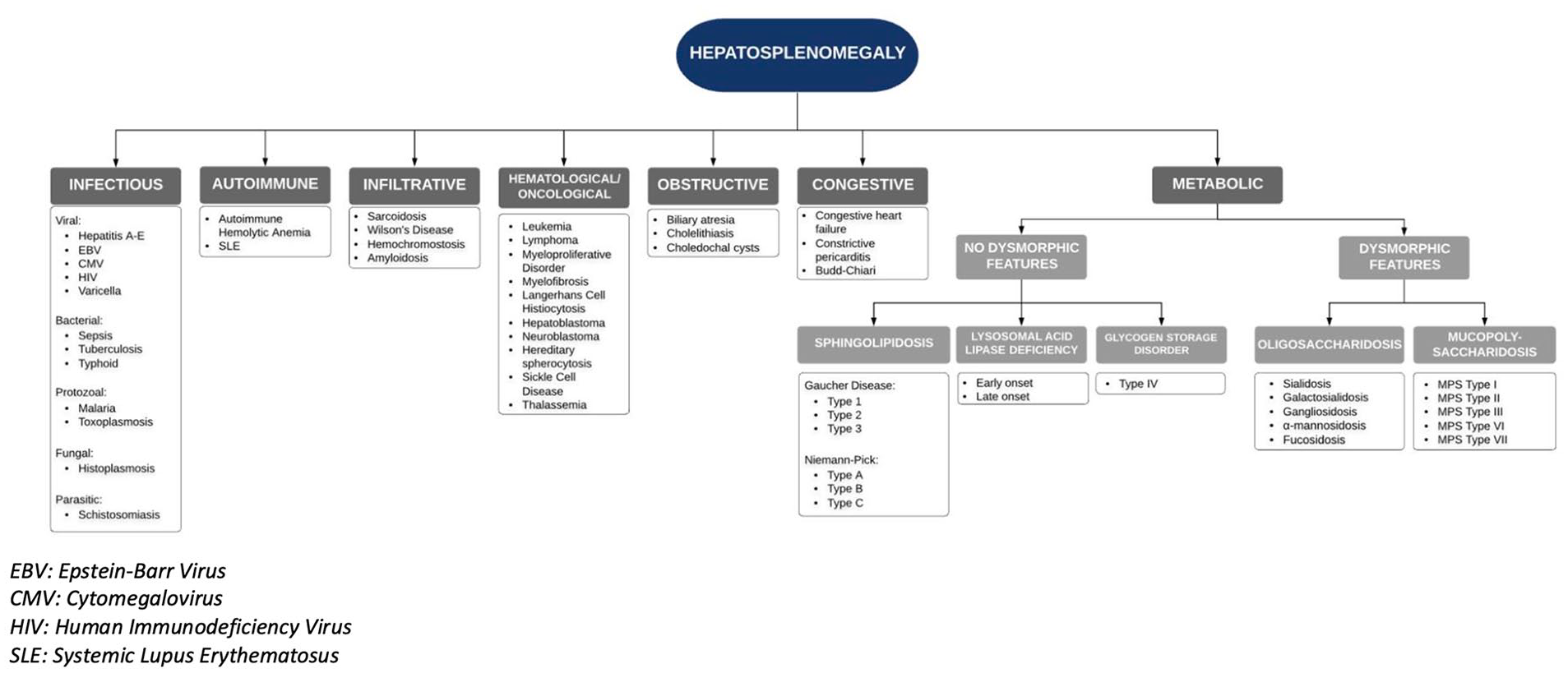
Approach to hepatosplenomegaly in pediatric patient.
As such, initial laboratory investigations were performed in the Paediatric Liver Clinic (Table 1). An infectious screen revealed a negative heterophile antibody test and nonreactive hepatitis A, B, and C serology. An immunological liver workup revealed normal immunoglobulins, C3 and C4, antinuclear antibody, anti-double stranded DNA, anti-MPO, anti-PR3, α-1-antitrypsin, and ceruloplasmin. Hematological investigations including hemoglobin electrophoresis, Coomb’s test, and G6PD deficiency screen were also negative. An α-fetoprotein level was also unremarkable. Metabolic storage disease was suspected, and testing revealed low leukocyte β-glucocerebrosidase activity and high plasma chitotriosidase activity (Table 1). In the context of hepatosplenomegaly, bony pain, easy bruising, and occasional epistaxis, we made a diagnosis of GD Type 1.
Laboratory Investigations.
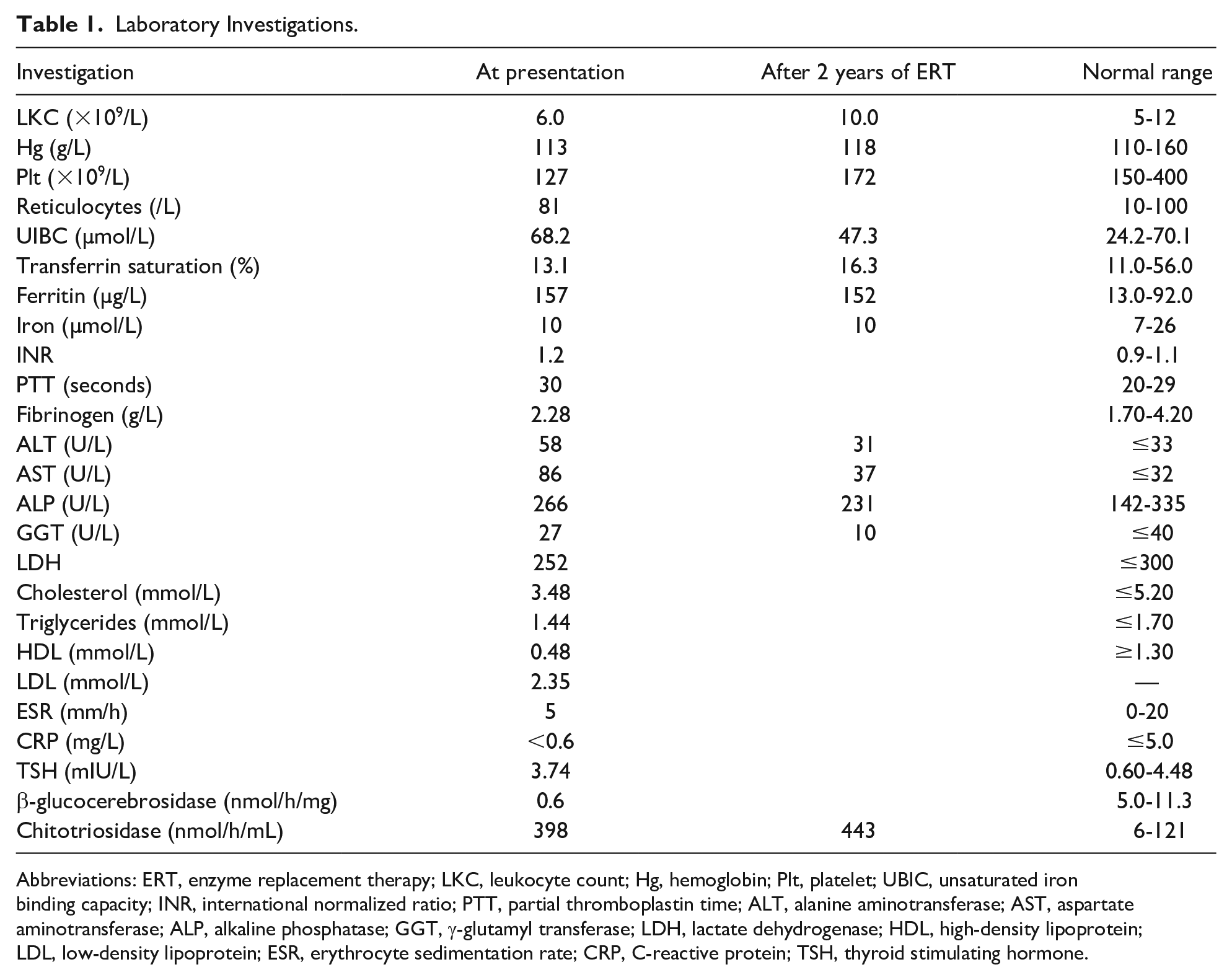
Abbreviations: ERT, enzyme replacement therapy; LKC, leukocyte count; Hg, hemoglobin; Plt, platelet; UBIC, unsaturated iron binding capacity; INR, international normalized ratio; PTT, partial thromboplastin time; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; GGT, γ-glutamyl transferase; LDH, lactate dehydrogenase; HDL, high-density lipoprotein; LDL, low-density lipoprotein; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; TSH, thyroid stimulating hormone.
The patient was referred to the Paediatric Metabolic Genetics team for further assessment. DNA sequencing of GBA identified 2 pathogenic variants, c.1226A>G and c.116_1505 deletion, confirming a diagnosis of GD Type 1. The c.1226A>G p.(N 409S; also known as N370S) is a common pathogenic variant. The c.116_1505 deletion is a novel deletion that spans exons 3 to 11 of GBA. A whole-body magnetic resonance imaging was performed to assess for bony abnormalities and identified mild femoral Erlenmeyer flask deformities bilaterally.
Final Diagnosis
Gaucher disease Type 1.
Course in Hospital
The patient was started on ERT with imiglucerase (Cerezyme) at 60 U/kg every 2 weeks. She tolerated this well with no reported side effects. Two years later, she continues to thrive with a weight of 28.4 kg (69th percentile) and height of 122.6 cm (15th percentile). Clinical examination and a repeat abdominal ultrasound demonstrated resolved hepatosplenomegaly with improved liver enzymes and thrombocytopenia (Table 1).
Discussion
Type 1 GD was first described in a doctoral thesis by Philippe Gaucher (1882) as a nonleukemic splenic epithelioma. 4 It is a rare condition with an estimated prevalence of 1 in 50 000 to 100 000 in the general population. 5 GD affects all ethnic and racial groups, through the prevalence is higher among those of Ashkenazi Jewish descent. The age of diagnosis is highly variable and typically correlates to the rate of substrate accumulation. 5 However, it is estimated that 66% of patients with GD Type 1 are diagnosed before 20 years of age. 6
In a longitudinal observational study, the most common signs and symptoms of GD in children included splenomegaly (95%), hepatomegaly (87%), bone disease (81%), thrombocytopenia (50%), anemia (40%), growth retardation (34%), and bone pain (27%). 7 Specifically, the 2 most common radiographic bone abnormalities in pediatric patients were Erlenmeyer flask deformities (49%) and bone marrow infiltration (38%). 7 Correspondingly, our patient was initially referred for hepatosplenomegaly and, after further investigation, was found to have concomitant thrombocytopenia and bilateral Erlenmeyer flask deformities of the femurs.
GD is diagnosed by measuring β-glucocerebrosidase activity in peripheral blood leukocytes. A finding of <15% of mean normal activity is supportive of the diagnosis. 8 Our patient had a reduced serum level of 0.6 nmol/h/mg (5.0-11.3 nmol/h/mg), which was diagnostic of GD Type 1. Her serum chitotriosidase, a marker expressed by chronically activated phagocytes and released by glucocerebroside-laden macrophages, was elevated at 398 nmol/h/mL (6.0-121 nmol/h/mL), further supporting a diagnosis of GD. 8
Molecular genetic testing confirms the diagnosis of GD and may provide valuable prognostic information. More than 300 variants of the GBA gene encoding for glucocerebrosidase have been associated with GD, with the most common being c.1226A>G p.N409S (N370S), c.84dupG p.L29fs (84GG), c.115+1G>A (IVS2+1G>A), and c.1448T>C p.L483P (L444P). 9 Genetic testing revealed that one of the patient’s mutations was the c.1226A>G (p.N409S) variant. This mutation is associated only with GD Type 1 and is thought to be protective against the development of the neurological involvement characteristic of GD Types 2 and 3. 9
ERT is the first-line treatment for GD Type 1 in pediatric patients. 10 ERT replaces the defective acid β-glucosidase and enables the catabolism of glucocerebroside, which accumulates in macrophages, into glucose and ceramide.10,11 Imiglucerase (Cerezyme) is most commonly used, though velaglucerase alfa (VPRIV) and taliglucerase alfa (Eyelyso) are also now approved for ERT in GD Type 1. The dose can range from 15 to 60 U/kg biweekly based on the clinical severity and response. There is reported improvement in hematological, visceral, and bone parameters within 6 months of therapy. 12 Other therapies include substrate reduction medications, bone marrow transplantation (used in some cases of types 2 and 3), splenectomy (no longer recommended), and potential gene therapies (currently being investigated in clinical trials).
Our patient was treated intravenously with 60 U/kg of imiglucerase (Cerezyme) biweekly. Her thrombocytopenia subsequently resolved with a platelet count of 170 × 109/L after 6 months and 172 × 109/L after 2 years of treatment. This is consistent with Weinreb et al’s study, where 54% of patients with initial platelet counts of 60 to 120 × 109/L reached normal levels after 24 months of ERT. 13 In this study, hemoglobin concentrations in anemic patients also increased to normal or near normal levels within 6 to 12 months. 13
Remarkably, at 2 years following initiation of ERT, our patient’s hepatosplenomegaly was undetectable by both clinical examination and ultrasound. This is also reflected in the literature, where liver volumes were found to decrease by 20% to 30% at 1 to 2 years, and 30% to 40% by 5 years of treatment. Splenic volumes similarly decreased by 30% within 1 year of therapy, and 50% after 2 years. 13 Finally, in patients with pretreatment bone pain, 52% were pain free after 2 years. 13 Although our patient still endorsed bony pain at a 2-year follow-up, this was clinically improved from initial assessment. A repeat magnetic resonance imaging 6 months following ERT initiation showed persistent mild Erlenmeyer flask deformities, but without bone infarcts.
A multidisciplinary approach is essential in the long-term management of GD Type 1. In addition to facilitating and observing response to ERT, medical specialists must monitor for long-term complications. In GD Type 1, aberrant macrophage activation and immune dysregulation are associated with an increased risk of cancer in the site of the metabolic disease and in secondary organs involved in the disease process. 14 Additionally, due to mutations in the GBA gene, it has been found that Parkinson disease may develop in 4% to 9% of GD Type 1 patients. 15 Long-term management should also encompass genetic counselling, as GD is inherited in an autosomal recessive manner. Last, allied health care professionals are vital in managing subsequent psychosocial aspects of care.
In summary, GD is a rare, but important, treatable diagnosis in pediatric patients presenting with hepatosplenomegaly. Associated hematological abnormalities including thrombocytopenia and anemia, and skeletal manifestations should raise clinical suspicion for GD. Measuring glucocerebrosidase activity in dried blood spots is a reliable and cost-effective method to diagnose GD early in individuals with hepatosplenomegaly and/or thrombocytopenia. Diagnosis is supported by a high serum chitotriosidase and genetic testing, with c.1226A>G being the most common deletion mutation seen in GD Type 1. Biweekly enzyme replacement therapy with imiglucerase has been proven to improve hematological, visceral, and bony manifestations of the disease. Physicians must consider the rare diagnosis of GD in patients presenting with hepatosplenomegaly to prompt early diagnosis and timely management.
Author Contributions
HCB: Contributed to conception and design; contributed to analysis; drafted the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
CP: Contributed to analysis and interpretation; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
CAP: Contributed to analysis and interpretation; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
DA: Contributed to analysis and interpretation; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Footnotes
Acknowledgements
We thank the patient and her family for allowing us to share her information.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.