
Abstract
Objective:
An aetiological link between acute infection and major depression has long been hypothesized, and is increasingly gaining recognition within contemporary literature. This review aims to examine the evidence for such a link, specifically between acute, self-limiting infection and major depression, and to summarize the current understanding of pathophysiological mechanisms underlying this link.
Methods:
Relevant articles were sourced via an online search of published literature from Embase, MEDLINE, PsycINFO and PubMed using a variety of search terms including mood disorder, depression, infection and inflammation. Additionally, a search for articles from the bibliographies of retrieved papers was conducted.
Results:
Findings from retrospective studies suggest an association between infection and subsequent mood disturbance, including major depression. This association has been confirmed by studies employing prospective observational or experimental challenge designs. The available evidence supports a multifactorial basis of vulnerability towards major depression in the context of acute infection. Genetic, neuroendocrine, autonomic and psychosocial factors may interact to potentiate the likelihood of a severe and prolonged depressive response to an immunological stressor in some individuals.
Conclusion:
Mood disturbance is likely to have a host-protective role in the context of an acute sickness response to infection. However, this usually adaptive and reversible response may progress in some vulnerable individuals into a more sustained and severe pattern of behavioural and physiological changes of major depression. Further research is needed to delineate the factors that predispose, precipitate and perpetuate depression in the context of acute infective illness. Such insights will inform effective prevention and treatment strategies.
Keywords
Introduction
Globally, more than 350 million people of all ages suffer from depression, which has been identified as a growing contributor to disability and the population disease burden worldwide (World Health Organization, 2012). Despite the immensity of this public health problem, research efforts to date have failed to identify a definitive aetiology for this condition, as it represents a spectrum of disorders (or subtypes) each with multiple predisposing (vulnerabilities), precipitating and perpetuating factors (American Psychiatric Association, 2000; Herald and Gordon, 2012).
A potential link between infection and mood disorder was speculated as far back as the 19th century, when Tuke (1892), in his Dictionary of Psychological Medicine, described 18 cases of post-influenza mania and depression admitted to the Bethlem Royal Hospital in London. It was not until almost a century later that a seminal paper detailing the behaviour of sick animals (Hart, 1988) provided the impetus for a scientific focus on the acute sickness response to infection. This research established that the physiological (e.g. fever), behavioural (e.g. anorexia, hyperalgesia, hypersomnolence) and emotional/motivational (e.g. depressed mood, anxiety, anhedonia, cognitive difficulties) changes that typically accompany significant infection are triggered by the action of immunological proteins (notably cytokines) on central nervous system (CNS) targets (Hart, 1988). In the past, physicians have generally regarded the acute sickness response as an unpleasant side effect of the necessary host immune response that occurs during active infection. However, these CNS-mediated changes are now understood to represent a conserved, neurobehavioural response to acute infection that works in tandem with the host immune response to facilitate pathogen clearance and recovery (Vollmer-Conna, 2001). Behavioural and emotional changes, including mood disturbance, are thus considered part of the normal response to acute infection. These changes are thought to serve the host primarily through preservation of energy to combat the pathogen (Goehler et al., 2007; Hart, 1988). Nevertheless, just as pathological fear and anxiety constitute debilitating variants of the fight-or-flight response, an excessive or prolonged sickness response can similarly be deleterious (Vollmer-Conna et al., 2004).
This article reviews the evidence for links between acute infection and mood disorder, and summarizes the current understanding of pathophysiological mechanisms underlying this link.
Scope of the review
The literature uses a range of terms to describe post-infective changes in mood, including mood disturbance, mood disorder, major depression and major depressive disorder (MDD). Steptoe (2007) suggested that this reflects an understanding of depression as a continuous variable, rather than a categorical entity. This is especially important in the context of medical illness, as even subthreshold disorders may lead to impairment. Consequently, Steptoe (2007) recommended that links between infection and depression would become clearer when the whole spectrum of depression is considered.
Somatic and psychological symptoms of mood disorder are divided in some studies. However, this is a problematic distinction in the context of immune activation, as the same cytokines that drive the somatic responses to infection are generally thought to also drive the psychological and neurobehavioural changes (Vollmer-Conna et al. 2004). Even outside the context of infection, a somatic symptom such as fatigue is described in the symptomatology of depression and included in the diagnostic criteria for major depression (American Psychiatric Association, 2000). When considering post-infective fatigue, White et al. (1998) suggested that when psychological symptoms are considered alone, they are more likely to indicate a psychological vulnerability, rather than representing post-infective depression. Hence, in this review, we have chosen to consider post-infective fatigue and other somatic symptoms as an overlapping condition with depression during and after an episode of acute infective illness in adulthood.
Evidence that mood disturbance can be triggered by infection
Retrospective studies
Multiple cross-sectional, case-control studies examining individuals identified after a previous infective episode have documented an apparent association between mood disturbance and prior infection with a range of agents. For example, an early study (Peszke and Mason, 1969) reported that among students visiting a university mental hygiene clinic, those with heterophile antibody titres suggestive of recent glandular fever were more likely to seek psychiatric help than those with titres below this value.
Multiple additional studies with cross-sectional designs have used immunoglobulin G (IgG) antibody titres against pathogens of interest to putatively implicate links between exposure to the pathogen and the presence or absence of psychiatric disturbances. For example, Thomas et al. (2004) found that seropositivity for Toxoplasma gondii or Coxiella burnetii (the bacterial causative agent of Q fever) in a population of English farmers was not associated with symptoms reflecting clinically significant psychiatric morbidity. However, this finding was limited by a lack of statistical power to test the study hypothesis as the prevalence of psychiatric symptoms was lower than expected in this relatively healthy occupational cohort. Similarly, Sinanan and Hillary (1981) found no significant difference in influenza antibody titres when comparing depressed and non-depressed patients. These studies are limited by the wide variation in the peak titres, and the kinetics of decline, in antibody levels in healthy subjects. In addition, the studies cannot account for exposure to other infectious agents, and often do not consider subthreshold levels of disturbed mood.
A more recent study (Soumahoro et al., 2009) reported that serological confirmation of Chikungunya virus infection (a mosquito-borne infection causing arthritis and constitutional symptoms), was linked with higher rates of self-reported depression during follow-up telephone interviews in the 17 months following infection compared to uninfected control subjects [relative risk (RR): 2.5; 95% confidence interval (CI): 1.5–4.1], and that the increased risk was largely attributable to prior infection with this pathogen (attributable risk percentage: 60%; 95% CI: 33.8–75.8%).
Similarly, Okusaga et al. (2011) found that seropositivity for influenza A, influenza B or coronaviruses was strongly associated with a history of major depression, as identified via the Structured Clinical Interview for DSM-IV Disorders, with the odds ratio (OR) for an association between major depression and seropositivity being 1.71 for influenza A (95% CI: 1.18–2.50), 2.65 for influenza B (95% CI: 1.65–4.25) and 2.72 for coronaviruses (95% CI: 1.88–3.94). This article also found that positive influenza B serology was significantly associated with a history of suicide attempt and a lifetime history of psychotic symptoms in patients with major depression.
In 1996, the Lancet published two letters describing protracted illness following acute Q fever (Ayres et al., 1996; Marmion et al., 1996), of which mood disturbance and fatigue were the major elements of the symptom complex. Subsequently, many studies have identified protracted fatigue and mood disorder to be associated with prior acute Q fever (Ayres et al., 1998; Hatchette et al., 2003; Marmion et al., 2005; Wildman et al., 2002). Specifically, Wildman et al. (2002) reported that during a 10-year follow-up of subjects after an outbreak of acute Q fever in the UK, psychological morbidity as assessed using the General Health Questionnaire was significantly more common in exposed subjects than in matched controls (46.8% vs. 23.4%). Similarly, Hatchette et al. (2003) found that 27 months after another outbreak of acute Q fever, infected participants had significantly lower scores on five of the eight scales in the Short Form (36) Health Survey, including the physical and mental health scales, compared to uninfected controls, thus reflecting impaired functional status.
In a population-based study of 10th graders in Norway, Lien et al. (2007) found a significant association between the report of two or more acute infections during the last 12 months and higher scores on indices of both internalized mental health problems (e.g. anxiety and depression as measured by the 10-item version of the Hopkins Symptom Checklist) and externalized (e.g. hyperactivity and conduct problems as measured by 10 items from the Strengths and Difficulties Questionnaire) mental health problems. More broadly, epidemiological studies have reported a highly replicated seasonal peak of suicide in spring (Postolache et al., 2010). Although multiple theories have been proposed, it is often noted that this peak follows or coincides with seasonal peaks in epidemics of upper respiratory viruses and related immune responses. Consistent with this hypothesis, Tonelli et al. (2008) reported that victims of suicide have an increased level of gene expression of allergy-related cytokines in the orbitofrontal cortex, a region of the brain displaying histopathological abnormalities in suicide victims. Similarly, rodent studies have reported that intranasal tree pollen administration in previously sensitized rats resulted in increased cytokine expression in the brain and behavioural changes consisting of increased anxiety and disturbed social interaction (Tonelli et al., 2009).
Taken together, the findings from these retrospective study designs generally support the notion that infection and depression are associated. The evidence is limited by the well-recognized shortcomings of such study designs, including biased reporting and retrospective attribution of events (Katz, 2001).
Prospective studies
More robust evidence supporting the phenomenon of post-infective depression has come from several prospective cohort studies examining infection outcomes to allow inference about the direction of causality between infection and mood disturbance. In an early study of 36 patients with infectious mononucleosis (IM) (also known as glandular fever), Cadie et al. (1976) found that in the 12 months following illness, rates of depression, as assessed by personal interview and the Middlesex Hospital Questionnaire, increased significantly in female patients (n = 20) from four cases prior to illness to 13 cases post infection. Interestingly, depression among male patients (n = 16) only rose by one additional case (from two to three cases), potentially implying a sex X pathogen interaction in risk.
In a series of papers, White et al. (1998; 2001) reported a prospective study of 250 patients from primary care who had presented with either glandular fever, or a simple upper respiratory tract infection (URTI). They reported that both infective illnesses were associated with depression during the acute phase of infection (Bruce-Jones et al., 1994). The RR of experiencing major depression during the acute infection was 6.9 (95% CI: 3.35–14.1), compared with the fortnight prior to the onset of infection. In addition, new episodes of major depression, lasting for up to 6 months, were found to be triggered in 28% of patients confirmed to have been infected with Epstein–Barr virus (EBV), in 14% of patients with non-EBV glandular fever and in 11% of those with URTI (White et al., 1998). These data suggest that the propensity to post-infective depression may vary between individual pathogens.
Buchwald and colleagues similarly studied a cohort of patients with IM caused by EBV infection, and found high levels of reported psychological distress during the early stages of the illness (Buchwald et al., 2000; Katon et al., 1999). The cohort indicated that failure to recover was evident in 38% of patients at 2 months and in 12% at 6 months (Buchwald et al., 2000). Although this report did not include psychiatric diagnoses, the reported symptoms, including decreased vitality and functional status, are suggestive of a fatigue syndrome or primary mood disorder. In a further paper from this cohort, Rea et al. (2001) reported that while most physical symptoms from the Symptom Checklist-90-revised had resolved by 1 month, fatigue and somnolence abated more slowly and were still commonly present at the 2- and 6-month assessments.
Smith et al. (1998) and Bucks et al. (2008) studied mood states after naturally occurring common colds. Smith et al. showed that infected participants felt significantly less alert and had lower hedonic tone (i.e. they were less happy and sociable) than the healthy controls. Similarly, Bucks et al. (2008) reported that this illness, even when patients were afebrile, significantly impaired mood. In both studies, increased overall symptom severity was associated with worsened mood. However, Smith et al. (1998) pointed out that since both measures were subjective, the correlations were likely to reflect individual differences in these ratings. Alternatively, when objective measures of symptom severity (e.g. mucus production) were measured, these did not correlate with mood – suggesting that mood and other components of the illness complex may have separate determinants.
We have previously reported the longitudinal course of illness after acute infection with EBV, Ross River Virus (RRV; a mosquito-borne infection that causes a rash and arthritis) or Q fever in the Dubbo Infection Outcomes Study (DIOS) (Hickie et al., 2006). In this prospective cohort, 27% of patients at 3 months, 12% at 6 months and 9% at 12 months continued to experience ongoing illness featuring fatigue and substantive disability. Although case rates for post-infective major depression were not reported in this analysis, symptoms of mood disturbance (notably irritability; rapidly changing moods, feeling unhappy or depressed) were prevalent in the acute phase, and had a slow course to resolution over months (Hickie et al., 2006).
Taken together, the evidence from these cohort studies suggested that multiple infective agents are implicated in triggering prolonged mood disorder, including both viral and bacterial pathogens. Moreover, various predictors of delayed convalescence have been identified. In a series of early studies, Imboden et al. (1959) reported that those with delayed convalescence (especially a propensity towards depression) following retrospectively studied acute brucellosis infection (a zoonotic bacterial pathogen), and prospectively studied Asian influenza (Imboden et al., 1961) showed greater premorbid emotional disturbance compared to those who recovered promptly. White et al. (2001) similarly reported that a premorbid psychiatric history (OR: 2.3; 95% CI: 1.4–3.9), an emotional personality (OR: 1.21; 95% CI: 1.11–1.35) and social adversity (OR: 1.7; 95% CI: 1.0–2.9) best predicted post-infective major depression, but not prolonged fatigue states. Post-infective fatigue lasting 6 months or more after the acute infection was instead predicted by a positive monospot test at onset (OR: 2.1; 95% CI: 1.4–3.3) and lower physical fitness (OR: 0.35; 95% CI: 0.15–0.8).
In the DIOS cohort we identified the severity of the initial illness as the strongest predictor of post-infective fatigue, whereas microbiological, demographic, socio-economic or psychiatric variables (personality characteristics, premorbid and/or intercurrent psychiatric disorder) were not predictive of prolonged fatigue (Hickie et al., 2006; Vollmer-Conna et al., 2008). These data suggest that while fatigue states and mood disorders may co-occur after infection, they appear to have distinct predisposing factors and hence potentially separate pathophysiologies.
Experimental and quasi-experimental studies
Experimental studies in both humans and animals have provided direct evidence supporting the aetiological association between an immunologically triggered acute sickness response and mood disturbance. Several studies have investigated the effects of an immunogenic stimulus in healthy volunteers.
Janicki-Deverts et al. (2007) and Smith et al. (1992) examined the relationship between mood and experimentally induced URTIs. The earlier studies (Smith et al., 1992) encompassed seven clinical trials and compared inoculation with five different viruses [rhinovirus (RV2, RV9), coronavirus (CV), respiratory syncytial virus (RSV) or influenza B] under the well-documented protocols of the Common Cold Unit (see Beare and Reed, 1977). Several findings were reported. First, the results failed to demonstrate a significant relationship between pre-challenge mood and susceptibility to infection and illness. Second, there was no indication that subclinical infection altered mood. However, results from the days when the volunteers were symptomatic, showed that the mood changes were dependent on the nature of the virus. Influenza B virus infections were associated with a general increase in negative affect, and CV-induced colds led to a reduction in alertness (but little change in anxiety or depression), while the other viruses produced no significant mood changes. Interestingly, altered mood was reported even after the primary symptoms of CV-induced URTIs had resolved. Janicki-Deverts et al. (2007) reported that local (nasal) production of the proinflammatory cytokines, interleukin (IL)-6, IL-1β or tumour necrosis factor alpha (TNF-α), was associated with reduced positive affect 24 h after experimental exposure to either influenza A or RV, but not placebo. Similarly, Reichenberg et al. (2001) and Eisenberger et al. (2009) both conducted double-blind, placebo-controlled studies with low-dose endotoxin administration and found significant increases in proinflammatory cytokine production and depressed mood in the endotoxin, but not the placebo, groups.
However, it was not clear from this work to what extent this decreased mood was a reaction to the physical illness, as opposed to being a direct result of the influence of proinflammatory cytokines on the CNS. Interestingly, studies that used a milder model of experimental immune activation, namely immunization against Salmonella typhi with the inactivated intramuscular vaccine Typhim Vi that contains endotoxin, found that even without physical symptoms or temperature increase, immunization led to a decline in mood over an 3-h period, in comparison with placebo (Harrison et al., 2009a, 2009b; Strike et al., 2004; Wright et al., 2005). Further, Wright et al. (2005) showed a significant correlation between the IL-6 response and subjective mood deterioration, with a similar trend reported by Harrison et al. (2009a).
A recent functional MRI study showed that mood deterioration in this typhoid immunization model was associated with enhanced activity within the subgenual anterior cingulate cortex (sACC) during emotional face processing (Harrison et al., 2009a). Additionally, the IL-6 response appeared to have a modulatory effect on the functional connectivity of the sACC to a network of regions implicated in mood regulation, namely the right amygdala, nucleus accumbens, medial prefrontal cortex and right superior temporal sulcus. This is significant, as the sACC is an important region implicated in the pathophysiology of major depression (Kumari, 2003; Mazheri et al., 2002). In fact, sACC activity has been shown to reverse with successful treatment of major depression, including with selective serotonin reuptake inhibitors (SSRIs) (Mayberg et al., 1999; 2000) and deep brain stimulation (Konarski et al., 2006; Mayberg et al., 2005).
Further insights are also available from studies of the effects of administration of recombinant human cytokines, used as a therapeutic intervention to boost immune responses in the treatment of certain cancers and hepatitis C infection (Dantzer et al., 2008). For instance, an influenza-like symptom complex including irritability is very common after the initial administration of interferon (IFN)-α therapy, and a significant minority of patients develop major depression and occasionally psychosis after more prolonged administration (Dantzer et al., 2008). Raison et al. (2006) reported that 50% of patients commencing high-dose IFN-α therapy met the criteria for major depression within 3 months. Far higher percentages (≥ 90%) develop at least one depressive symptom (Capuron et al., 2002a). Moreover, increases in IL-6 and TNF-α production during therapy were shown to correlate with increases in depressive symptoms (Raison et al., 2010).
An extensive body of evidence from animal studies has produced findings consistent with the observations in humans that activation of the immune system can cause depression. Anhedonia, as assessed by decreased preference for sweetened drinking solutions (Frenois et al., 2007; Weil et al., 2006), and reduced intracranial self-stimulation (Barr et al., 2003; Borowski et al., 1998), were reported in endotoxin or cytokine-treated mice. This model involves reinforcing electrical stimulation of the forebrain bundle at the level of the posterior lateral hypothalamus and does not involve appetite regulation, and is therefore considered a more pure rodent model of anhedonia (Liebman, 1983). Other classical depression-like behaviours, including decreased social and explorative behaviours (Larson and Dunn, 2001; Stone et al., 2006), and helplessness in inescapable situations as assessed by increased immobility in the tail suspension test and in the forced swim test (Frenois et al., 2007), were also reported after endotoxin or cytokine administration. Furthermore, Frenois et al. (2007) reported that these behaviours persisted even after motor activity, food intake and drinking returned to normal, suggesting that the mood changes were determined independently. It should be noted, however, that the dosages of cytokines administered in these studies were supraphysiological, and that in the human studies the observed effects may be confounded by the underlying medical illness (Vollmer-Conna, 2001).
Further evidence for the notion that infection-associated mood disorder has common features with that arising without an infective trigger is provided by the beneficial effects of pretreatment with antidepressant drugs on the depressive symptoms in cytokine therapy (Musselman et al., 2001). Similarly in animal models, antidepressant drugs reversed the reduced consumption of a sweetened solution in endotoxin-treated rats (Yirmiya et al., 1999), and enhanced the performance of IL-1-treated rats in sucrose solution-reward tasks (Merali et al., 2003). There is evidence showing that in addition to their central effects, antidepressant drugs may also act on peripheral immune responses directly (Baganz and Blakely, 2013; Frick and Rapanelli, 2013). SSRIs, for example, increase serotonin levels in the periphery as well as in the brain. The peripheral effect is thought to play a role in enhancing the cytolytic function of natural killer cells and increasing B-cell proliferation. SSRI treatment has also been shown to normalize elevated cytokine levels in patients with depression; however, this effect on cytokine levels was linked to a therapeutic response to the antidepressant drug (Baganz and Blakely, 2013).
Putative mechanisms underlying immunologically triggered mood disorders
It is now understood that the host response to infection requires synchrony between metabolic, physiological and behavioural components. For example, the conservation of energy that occurs through decreased activity levels during sickness is thought to support the increased demand for energy associated with raising body temperature during the fever response and other aspects of the host response. Although the fever response is costly in terms of energy, its adaptive nature is well documented from numerous demonstrations that the inhibition of fever (e.g. by placing infected animals in a cold environment, or treating them with antipyretic drugs) is detrimental to survival (Kluger, 1979). Interference with other aspects of the sickness response, for example, by force-feeding or sleep depriving animals with acute infections, similarly produced a marked reduction in survival rates (Hart, 1988; Kluger, 1979).
The behavioural and physiological changes during sickness are additionally thought to facilitate recuperative vegetative states, and to provide social benefits, such as signalling the need for help and a reduction in competitive behaviours by others (Hart, 1988; Vollmer-Conna, 2001). Thus, the sickness response is not merely an automatic, inflexible reaction to illness, but rather an expression of a central motivational state that functions to reorganize behavioural priorities during sickness (Dantzer, 2001; Vollmer-Conna, 2001).
Raison et al. (2006) proposed that the presence of mood disturbance as a requisite behavioural component of immune activation, suggests that it is pleiotropically linked to genes that directly, or indirectly, promote enhanced inflammatory responses. Such allelic variants, despite posing a risk of depression, might have concurrently improved overall fitness by increasing resistance to pathogen assault before the advent of modern sanitation and antibiotic therapy. The physiological and behavioural changes that characterize the acute sickness response are well documented in a wide range of vertebrate species, indicating strong evolutionary conservation of the phenomenon (Hart, 1988; Konsman et al., 2002).
Thus, mood disturbance appears to be linked to the overall response to infection. This does not imply that depressive illness is adaptive – except in the temporary context of acute infection. In healthy individuals, this evolutionarily conserved mechanism allows cytokines to orchestrate a highly adaptive and reversible behavioural response to infection. However, in vulnerable individuals, this acute response to infection may trigger a sustained pattern of physiological, biochemical and behavioural perturbations that manifest as the symptoms of depression (Dantzer et al., 2008).
Individual differences in vulnerability are clearly evident despite the relatively stereotyped set of symptoms and behaviours of the acute sickness response. Even where there is infection with invariant pathogens, significant variability exists in both the severity of the symptom complex experienced by individuals, and the detailed characteristics of the illness complex (Piraino et al., 2012; Vollmer-Conna, 2001; Vollmer-Conna et al., 2008). We recently reported that the illness complex in both the acute phase and the protracted course, features relatively discrete symptom domains or endophenotypes, including mood disturbance, pain and fatigue, which are temporally stable within individual subjects and have unique genetic determinants (Piraino et al., 2012). Accordingly, some individuals report prominent fatigue, while others experience prominent mood disturbance, cognitive difficulties or pain. In some, the mood disturbance may be relatively mild whereas others may develop a major depressive episode with suicidal ideation (Piraino et al., 2012; Raison et al., 2006).
In considering the wide variability in the response to infection, it is important to recognize that normal recovery requires a complex sequence of activation and inhibition, not only in immune pathways but also in other systems involved in the maintenance of homeostasis and body integrity. Pre-existing (inherited or acquired) vulnerabilities in one or more response systems may underpin the individual illness manifestations. In the context of IFN-α therapy, the extent of hypothalamic–pituitary–adrenal (HPA) axis activation in response to the initial injection of IFN-α was strongly associated with the subsequent development of major depression; and this was hypothesized to reflect a pre-existing sensitization of corticotropin-releasing factor pathways (Capuron et al., 2003).
Thus, resilient individuals may be protected from developing depression in response to all but the highest levels of inflammatory stimulation (explaining the high rates of sickness symptoms, including mood disturbance, seen with supraphysiological doses of cytokine therapy), whereas others may develop depressive symptoms in response to even low levels of immune stimulation. A predisposition towards depression during the acute sickness response to infection may be further potentiated by concurrent psychosocial stressors, which add to the overall vulnerability of an individual (Cohen et al., 1999).
In recent years, renewed scientific interest has focused on the potential importance of circadian functioning, and of sleep in particular, in mood regulation (for a review, see Harvey, 2011). Various types of sleep disturbance are comorbid with mood disorders. Insomnia and hypersomnia are listed in the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) as symptoms of MDD (American Psychiatric Association, 2000). Similar to the current understanding of the acute sickness response, there has been a shift away from conceptualizing sleep disturbance as merely epiphenomenal to mood disorder. Instead, it has been proposed that sleep disturbance is an important mechanism contributing to the development and maintenance of mood disorders (Harvey, 2008).
The acute sickness response to infection is characterized by changes in sleep behaviour mediated by alterations in the levels of proinflammatory cytokines (Hart, 1988). A large body of evidence derived from electrophysiological, biochemical, molecular and genetic studies have established a role for IL-1β and TNF-α in the regulation of spontaneous sleep–wake behaviour (reviewed in Opp, 2005). Activation of these cytokine systems during an acute immune response to infection results in an increased need to sleep and a sleep phenotype that is characterized by enhanced duration of non-rapid eye movement sleep accompanied by slow-wave electroencephalographic activity (or delta power) indicating deep sleep. Cytokines implicated in modulation of sleep–wake behaviour have been shown to exert their effects via several related neurotransmitter, peptide or hormone systems (Besedovsky et al., 2012; Opp, 2005). While changes in sleep pressure, duration and intensity have been well documented (as they occur during the acute phase of an infective illness), data on possible longer-term alterations in sleep and how these may interact with post-infective neuropsychiatric syndromes are lacking. There is, however, increasing evidence for a reciprocal relationship between sleep and immune functioning, and a growing understanding that poor and disordered sleep can have detrimental effects on the immune responses (Besedovsky et al, 2012; Bryant et al 2004).
A series of animal experiments have recently documented that imbalance of autonomic neural activity during an acute infection can significantly affect the intensity of the inflammatory response, symptom severity, survival or length of recovery from the infection (Borovikova et al., 2000; Grebe et al., 2010; Pavlov et al., 2009; Thayer and Sternberg, 2010). Whether or not altered circadian rhythmicity in autonomic or other systems is instrumental to a failure to recover and the development of post-infective syndromes including depression is not as yet known.
Biological factors associated with the acute sickness phenotype of depressed mood
Genetic variation
The importance of genetic contributions to the manifestations of the acute sickness response to infection was initially indicated by the seminal research of the Danish Adoption Register (Sørensen et al., 1988), which identified a 5.8 RR of an adopted child dying from infection, if the biological parent has also died of infection before age 50. In comparison, the RRs for cancer and cardiovascular death were 1.2 and 4.5, respectively.
More recently, we reported that functional polymorphisms in cytokine genes with important roles early in the immune response, notably IFN-γ and IL-10, were significantly associated with proinflammatory cytokine production, as well as the course of illness following EBV, RRV or Q fever infection (Vollmer-Conna et al., 2008). Specifically, those individuals whose genetic make-up favoured a more intense inflammatory reaction experienced more severe symptoms of sickness and a more protracted illness. As described previously, individuals differ in the relative contributions of individual symptom domains or endophenotypes making up the illness complex (Piraino et al., 2012). Individuals who experienced mood disturbance as the most significant aspect of the sickness response, were more likely to have a genotype that resulted in higher levels of the proinflammatory cytokine IL-6, and lower levels of the anti-inflammatory cytokine IL-10.
Genetic polymorphisms within the serotonergic system [notably, the serotonin transporter gene solute carrier family 6 (neurotransmitter transporter), member 4 (SLC6A4) have also been associated with vulnerability to the development of depression after IFN-α therapy (Bull et al., 2009; Kraus et al., 2007; Lotrich et al., 2009). Similarly, SLC6A4 polymorphisms have been associated with vulnerability to tryptophan depletion (Neumeister et al., 2002), raising the possibility that these genetically vulnerable subjects may benefit from SSRI prophylaxis during IFN-α therapy (Lotrich, 2009).
Neurotransmitters and biochemical pathways
The acute sickness response to infection is initiated by the production of proinflammatory cytokines, but is ultimately mediated by neurochemical changes within the brain. Thus, the interactions between cytokines and neurotransmitters need to be considered. Cytokine-related alterations in the metabolism of the neurotransmitters serotonin, norepinephrine (noradrenaline) and dopamine in the limbic system (a brain region essential to the regulation of emotion and motivation) and the basal ganglia (involved in the regulation of psychomotor function and reward) have been reported (Dunn et al., 1999; Gao et al., 2002).
There has been a growing interest in a possible role for the bioavailability of tryptophan (an amino acid essential for the synthesis of serotonin) in infection/inflammation-associated depression (Dantzer et al., 2008; Konsman et al., 2002). As shown in Figure 1, tryptophan is metabolized down one of two pathways – the serotonin pathway or the kynurenine pathway. Major enzymes involved in the kynurenine pathway, such as tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO) are elevated during immune activation (Konsman et al., 2002). IDO, which normally plays a negligible role in tryptophan metabolism, is directly activated by a number of proinflammatory cytokines, including IFN-α and TNF-α. Similarly, TDO activation is induced by glucocorticoids, and as cytokines can activate the HPA axis, TDO activity may also increase. Together, these actions potentiate the kynurenine pathway and decrease tryptophan availability for serotonin synthesis. In fact, cytokine immunotherapy studies show marked reduction in the plasma levels of tryptophan (Dantzer et al., 2008; Konsman et al., 2002). In combination with pre-existing vulnerabilities, the downstream effects of reduced serotonin bioavailability may precipitate mood disorder during an acute sickness response to infection.
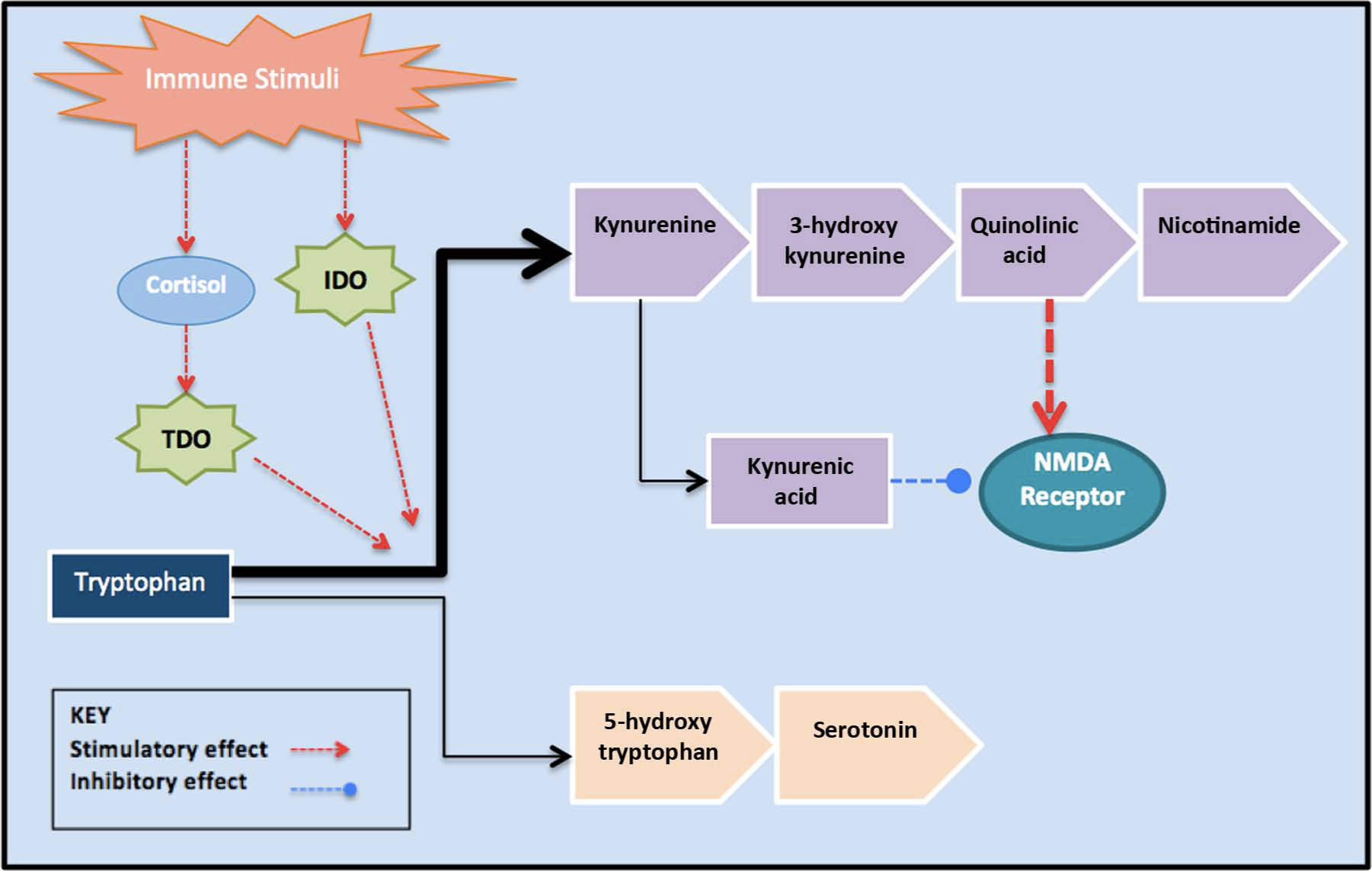
The effects of the immune stimuli on tryptophan metabolism. The major enzymes involved in the kynurenine pathway, such as tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO), are elevated during the inflammatory response. This increases kynurenine pathway activity and reduces tryptophan availability for serotonin synthesis. The neuroactive kynurenine pathway metabolites are thought to act on the N-methyl-
Recent evidence suggests a role of heightened glutamate receptor activation in major depression (Müller and Schwarz, 2007). Increased kyurenine pathway activity may contribute to this, as kyurenine itself can be transported across the blood–brain barrier for further metabolism to generate neuroactive compounds that stimulate glutamatergic activity (Dantzer et al., 2008). While both agonists (e.g. quinolinic acid) and antagonists (e.g. kynurenic acid) of glutamate receptors are generated via this pathway, the net effect is thought to favour glutamatergic hyperfunction (Müller and Schwarz, 2007).
Both clinical and experimental data support the likely involvement of altered tryptophan and IDO availability in immune-associated depressive disorders. In fact, a landmark paper by Capuron et al. (2002b) demonstrated that immune activation through repeated IL-2 and IFN-α immunotherapy in cancer patients induced a drastic fall in plasma tryptophan levels, which was positively correlated with depression scores. It should be noted, however, that these mediators are unlikely to be the sole mechanism, as proinflammatory cytokines have also been shown to increase tryptophan uptake in the brain and enhance serotonin turnover (Dunn et al., 2005).
Growth factors, notably brain-derived neurotrophic factor (BDNF), with documented roles in neurogenesis and neuroplasticity, have also been implicated with post-infective mood disturbance. Inflammatory cytokines were found to disrupt BDNF signalling by decreasing BDNF release and by interfering with BDNF receptor phosphorylation (Cortese et al., 2011; Felger and Lotrich 2013). Consistent with this, immune activation (via IFN-α administration or lipopolysaccharide injections) has been shown to decrease systemic BDNF levels (Cortese et al., 2011; Lotrich et al., 2013). Similarly, gene association studies have documented a specific link between the low-producing Met allele of the BDNF Val66Met polymorphism and the psychological, but not neurovegetative, symptoms of depression in the context of IFN-α treatment (Lotrich et al., 2013). However, it has also been shown that during IFN-α administration, BDNF decreased regardless of whether an individual developed depression or not (Felger and Lotrich, 2013), which is consistent with findings from animal experiments showing that IFN-α can affect behaviour without affecting BDNF levels (Fahey et al., 2007).
In view of these seemingly incongruous findings, it is likely that the risk for cytokine-induced depression is particularly elevated in individuals with pre-existing (genetic or acquired) vulnerabilities towards low levels of BDNF. This premise is consistent with the reported risk associations between depression and the Met allele of the BDNF Val66Met polymorphism in other contexts, and further supports the notion that specific genetic variations and neurochemical systems may confer either resilience or susceptibility to manifestations of depression when threatened by an immunological stressor (Felger and Lotrich, 2013).
Stress response pathways
The proinflammatory cytokines produced during the acute sickness response are likely to affect neuroendocrine pathways (Papanicolaou, 1998), including the stimulatory effects on HPA axis hormones, especially corticotropin-releasing hormone (CRH) in the hypothalamus and the amygdala, a brain region associated with fear and anxiety (Besedovsky and Del Rey, 1996; Capuron and Miller, 2004; Silverman et al., 2005). In addition, proinflammatory cytokines can disrupt glucocorticoid receptor signalling (McKay and Cidlowski, 1999) and thus contribute to alterations in glucocorticoid-mediated feedback regulation of both CRH and thus further proinflammatory cytokine release.
The experimental administration of proinflammatory cytokines or endotoxin in rodents has revealed that along with the acute sickness response, a classical stress response is produced (Maier and Watkins, 1998; 1999; Raison et al., 2006). This was portrayed by the activation of the sympathetic nervous system, release of plasma catecholamines and activation of the HPA system, leading to the release of adrenocorticotropic hormone and glucocorticoids (Maier and Watkins, 1998).
The similarities between aspects of the stress response and acute sickness response point to a common mediator. It is generally regarded that the systems mediating host defence against infection on the one hand, and against psychosocial and environmental stressors on the other, are closely connected (Vollmer-Conna, 2001). It has been proposed that environmental (i.e. external) stressors activate the similar neuro–endocrine–immune circuitry as immunogenic agents (i.e. internal stressors), although they may enter the circuit at different sites (Maier and Watkins, 1998; 1999). Thus, differences in the sensitivity of stress response systems are likely to be associated with variation in illness manifestations during infection. Supporting this view, Raison and Miller (2011) recently reported that increased inflammatory responses to a laboratory psychosocial stress test administered prior to commencing IFN-α therapy predicted increased depression several months later during IFN-α therapy.
The autonomic nervous system (ANS) is increasingly understood to play an important role, not only in vegetative regulation, but in modulating the intensity of the host response to immunogenic stimuli (Grebe et al., 2010; Thayer and Sternberg, 2010; Tracey, 2009). For example, alpha-adrenergic sympathetic activation in response to a significant immunogenic challenge, such as a potentially lethal dose of influenza A virus, was recently shown to critically impact on the innate immune response and to increase morbidity and mortality (Grebe et al., 2010). On the other hand, stress-related sympathetic activation can also engender a set of changes similar to the host response to acute infections, including the secretion of proinflammatory cytokines, increases in temperature and cardiovascular changes (Carrive, 2006; Gibb et al., 2008; Kadota et al., 2010; Maier, 2003; Maier and Watkins, 1999). Substantive sympathetic reactivity during a severe, acute immune challenge may thus result in an overly vigorous response and increase symptoms of sickness including mood disturbance.
The potential role of the parasympathetic (vagal) branch of the ANS during immune activation has only recently become the focus of scientific interest. This research has revealed the potential of the cholinergic anti-inflammatory pathway in containing inflammatory responses (Borovikova et al., 2000; Pavlov et al., 2009; Thayer and Sternberg, 2010; Tracey, 2009). The acute sickness response includes components suggestive of vagal dominance – in particular increased slow-wave sleep – which is understood to be important in promoting recovery (Goehler et al., 2007; Rahman et al., 2011). No studies of the potential role for autonomic modulation of the mood component of the acute sickness response have been conducted to date.
Psychosocial factors in mood disturbance
A body of literature underscores the potential importance of psychosocial factors and personality in the development and maintenance of severe mood disturbance. Personality style, notably neuroticism, is well documented to shape an individual’s response to stressors (including illness) and to inform the selection of strategies for coping with adversity (Coulston et al., 2013; Ormel et al., 2013). In relation to the current context, a high score on the trait of emotionality (neuroticism) was associated with prolonged mood disturbance after an acute viral infection in two prospective cohort studies (Hickie et al., 2006; White et al., 2001), and a previous psychiatric history in combination with high neuroticism scores predicted a major depressive episode after infection with EBV (White et al., 2001).
Social adversity was also found to play an important role in perpetuating mood disorder in patients whose depression lasted from 2 to 6 months following an acute illness (Bruce-Jones et al., 1994). Those with a psychiatric diagnosis at the 2- and 6-month follow-up were 9.1 and 11.9 times more likely to have experienced significant stressful events during the follow-up period compared with those without psychiatric diagnoses at these two follow-up assessments. Two further studies (Buchwald et al., 2000; Katon et al., 1999) reported similar trends, stating that although both biological and psychosocial factors are highly correlated with psychological distress at 2 months, perpetuating psychosocial factors were more important at the 6-month follow-up.
Similarly, Raison et al. (2005) observed that even mild increases in depressive and anxiety symptoms just prior to treatment strongly predicted the development of depression during IFN-α therapy. Buchwald et al. (2000) suggested that adverse life experiences may have a cumulative effect, overwhelming a vulnerable person’s psychological and physiological resources. Similarly, past mood disorders or a neurotic personality type may serve as a marker for existing psychopathology, or a stress response system that is already vulnerable, thus predisposing some individuals to more severe mood disturbance in response to a significant illness stressor. A limitation of many studies exploring a role for psychosocial factors in mood disturbance associated with acute infection is that they did not investigate biological factors; hence, it can be argued that the psychosocial risk factors identified may actually reflect underpinning biological vulnerabilities including genetic predispositions.
Summary and conclusion
Many studies have reported that mood disturbance follows from acute infective illnesses due to a range of pathogens. Although mood disturbance is recognized to be a normal component of the acute sickness response to infection, severe or prolonged depression can be debilitating. Genetic, neuroendocrine, autonomic and psychosocial factors (Table 1) may interact to potentiate the likelihood of a severe depressive response to an immunogenic stressor in vulnerable individuals. Further studies are needed to delineate the factors that predispose, precipitate and perpetuate depression in the context of acute infective illness. A better understanding of the mechanisms critical in the evolution and perpetuation of this debilitating condition will also inform effective preventative and treatment strategies.
Summary of the putative mechanisms underlying the manifestations of immunologically triggered sickness response, including mood disturbance.
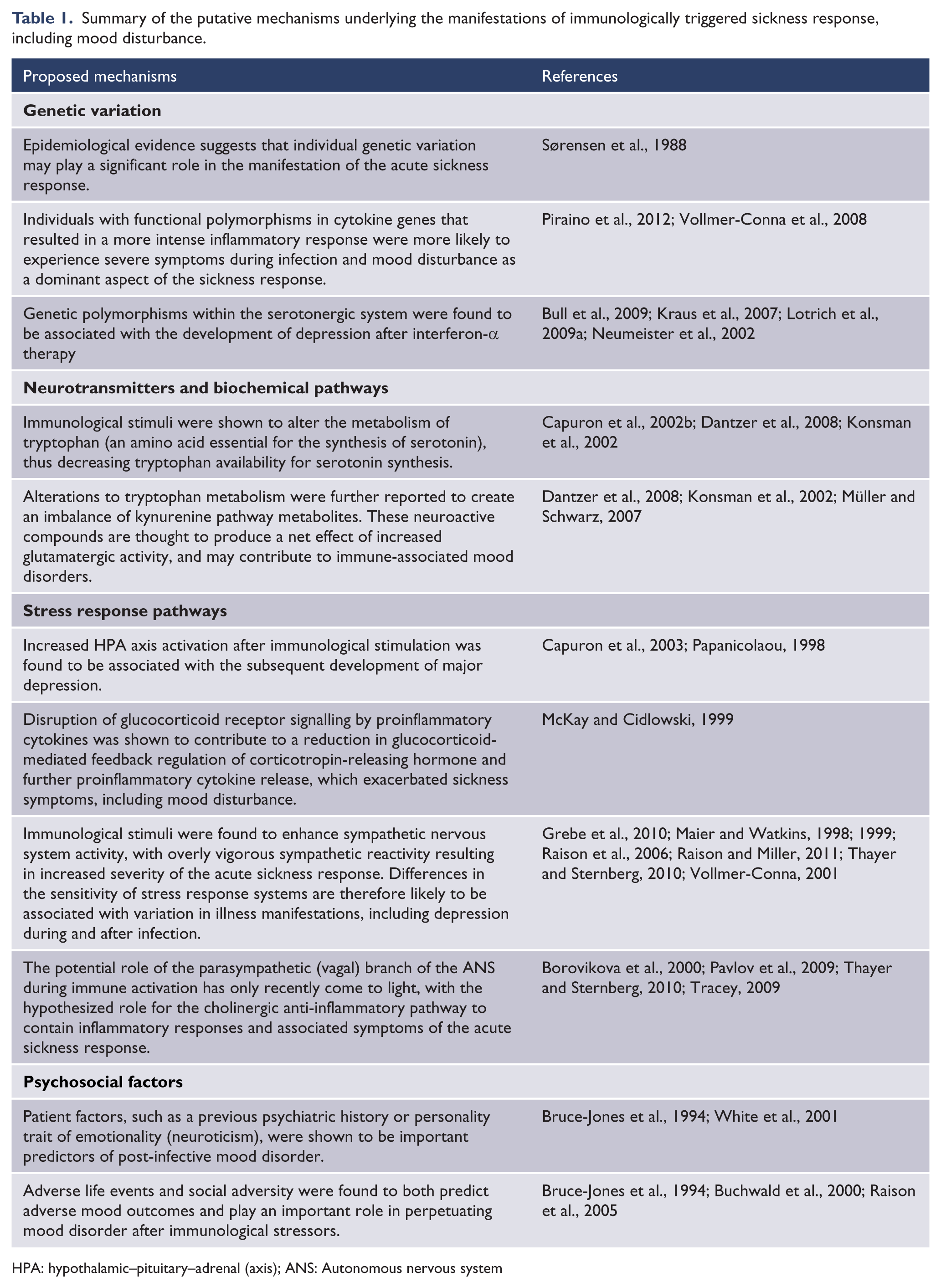
HPA: hypothalamic–pituitary–adrenal (axis); ANS: Autonomous nervous system
Footnotes
Acknowledgements
We thank Professor Ian Hickie for his helpful suggestions in preparing the final version of this manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.