
Abstract
Objectives:
Bipolar patients frequently relapse within 12 months of their previous mood episode, even in the context of adequate treatment, suggesting that better continuation and maintenance treatments are needed. Based on recent research of the pathophysiology of bipolar disorder, we review the evidence for mitochondrial dysregulation and selected mitochondrial modulators (MM) as potential treatments.
Methods:
We reviewed the literature about mitochondrial dysfunction and potential MMs worthy of study that could improve the course of bipolar disorder, reduce subsyndromal symptoms, and prevent subsequent mood episodes.
Results:
MM treatment targets mitochondrial dysfunction, oxidative stress, altered brain energy metabolism and the dysregulation of multiple mitochondrial genes in patients with bipolar disorder. Several tolerable and readily available candidates include N-acetyl-cysteine (NAC), acetyl-L-carnitine (ALCAR), S-adenosylmethionine (SAMe), coenzyme Q10 (CoQ10), alpha-lipoic acid (ALA), creatine monohydrate (CM), and melatonin. The specific metabolic pathways by which these MMs may improve the symptoms of bipolar disorder are discussed and combinations of selected MMs could be of interest as well.
Conclusions:
Convergent data implicate mitochondrial dysfunction as an important component of the pathophysiology of bipolar disorder. Clinical trials of individual MMs as well as combinations are warranted.
Introduction
Patients with bipolar disorder frequently experience multiple episodes of depression, mania, or hypomania, even if treated with the best available pharmacotherapy. Prospective longitudinal studies suggest that relapse rates within a year approach 67% for a cohort of bipolar patients with mostly inpatient index episodes (Judd et al., 2008). The latest effectiveness trial information from the STEP-BD study showed about 50% of bipolar patients will relapse within 12 months of recovering from an outpatient index mood episode (Perlis et al., 2006). Better continuation and maintenance treatments are needed.
We review potential mitochondrial modulators (MM) for the treatment of bipolar disorder that builds on recent findings on the pathophysiology of bipolar disorder. We focus on: (a) mitochondrial dysfunction in bipolar disorder (Clay et al., 2011; Konradi et al., 2004; Munakata et al., 2004, 2007; Naydenov et al., 2007; Quiroz et al., 2008; Shao et al., 2008b; Stork and Renshaw, 2005; Wang, 2007); (b) altered brain energy metabolism (Frey et al., 2007a); and (c) an important convergent genetic finding about the alpha-1C subunit of the L-type calcium channel (Sklar et al., 2008) along with calcium channel physiology associated with bipolar disorder, mitochondrial function and apoptosis. While each MM has the potential for treating bipolar disorder, it is possible that a combination of MMs (which target multiple points in the mitochondrial electron transport chain as well as oxidative stress) added to standard mood stabilizer treatment could reduce bipolar relapse or resurgence of mood symptoms. The purpose of this paper is to review the spectrum of MMs that could be considered for clinical trials. Of note, all MMs reviewed here are endogenous substances, currently available commercially as ‘nutraceuticals’ or dietary supplements.
Background
Bipolar disorder, with a prevalence of about 1% for BPI and 1.1% for BPII, is associated with substantial lifelong disease burden and dysfunction and tends to be highly recurrent (Merikangas et al., 2007; Weissman et al., 1996). The NIMH Collaborative Depression Study followed a group of unipolar and bipolar patients for almost 25 years. Of the bipolar patients, most of who were recruited from inpatient settings, almost 67% experienced at least one relapse within a year of recovery, with a higher risk of relapse if they experienced residual mood symptoms (Judd et al., 2008). A subset of bipolar participants in STEP-BD, who were followed prospectively after recovery from a mood episode, had recurrence rates of about 50% within 12 months, despite measurement-based, guideline-informed, systematic care with a fully available complement of modern psychopharmacological treatments – and these participants also had higher relapse rates if they had residual manic symptoms (Perlis et al., 2006). Furthermore, subsyndromal symptoms of depression, mania and hypomania with associated dysfunction frequently persist between episodes (Judd et al., 2008). Better treatments are needed to keep bipolar patients well, reduce subsyndromal symptoms, and prevent subsequent mood episodes.
Because the pathophysiology of bipolar disorder has yet to be determined, developing rational novel therapies presents a formidable challenge (Mathew et al., 2008). Exciting new research that converges on mitochondrial dysfunction, however, could lead to a new drug development paradigm for the treatment of bipolar disorder (Kato, 2008; Kazuno et al., 2008; Munakata et al., 2004, 2007). We postulate that MMs are candidates for novel treatments based on the latest studies of mitochondrial abnormalities (Clay et al., 2011) and alterations of brain energy metabolism (Frey et al., 2007a; Stork and Renshaw, 2005) in bipolar disorder, as well as an important whole genome-wide association study that shows altered single nucleotide polymorphism (SNP) for L-type calcium channels (Sklar et al., 2008). The strength of the approach is that it can address a range of dysfunctional mitochondrial targets, especially if combinations of MMs are used. It is possible that the effects of several MMs could be synergistic or target different abnormalities across the electron transport chain within mitochondria; alternatively, using several modulators may not be any better than using one. The potential use of multiple MMs is also consistent with the need for rational polypharmacy in bipolar patients and the finding that participants in STEP-BD took a median of three medications (Ghaemi et al., 2006).
Mitochondrial dysfunction in bipolar disorder
Multiple lines of evidence converge to strongly implicate mitochondrial dysfunction and oxidative stress in bipolar disorder (Clay et al., 2011). While the degree of mitochondrial dysfunction may not be severe enough to manifest a systemic disorder, it may be enough to cause neuropsychiatric symptoms as the brain requires significantly more energy than any other organ in the body (Peters et al., 2004). Mitochondria regulate energy production and generation of adenosine-5′-triphosphate (ATP) through the mitochondrial electron transport chain (ETC), with associated production of reactive oxygen species (ROS) that can result in oxidative stress and cellular damage, especially in the absence of sufficient antioxidant defenses (Ng et al., 2008). In addition to oxidative processes, mitochondria regulate calcium and apoptotic processes, and are central to facilitating neuronal plasticity. Dysfunctional mitochondria can result in neuronal damage via multiple mechanisms: decreased ATP production, oxidative damage of membranes and DNA, abnormal calcium sequestration, and apoptosis via activation of caspase proteases (Ng et al., 2008; Wang, 2007). As reviewed below, mitochondrial dysfunction in bipolar disorder is associated with decreased ATP production, indicated by decreased brain energy metabolism and a shift from oxidative phosphorylation to anaerobic glycolysis (Cui et al., 2007; Frey et al., 2007a; Naydenov et al., 2007; Stork and Renshaw, 2005) (as shown by decreased phosphocreatine (PCr) and ATP in magnetic resonance spectroscopy (MRS) studies), upregulation of genes involved in apoptosis compared to patients with schizophrenia (Benes et al., 2006), downregulation of mitochondrial genes regulating OXPHOS and proteasome degradation in bipolar disorder as compared to those with schizophrenia and healthy controls (Konradi et al., 2004), decreased antioxidant defenses, including decreased superoxide dismutase and glutathione-peroxidase-1 and -4 (Konradi et al., 2004), and increased lipid peroxidation, as well as abnormal calcium metabolism (Kato, 2008; Munakata et al., 2004). Moreover, abnormalities in the structure and distribution of mitochondria have been identified in brain and peripheral cells obtained from patients with bipolar disorder (Cataldo et al., 2010). In summary, substantial evidence supports the hypothesis that mitochondrial dysfunction may play a central role in the pathophysiology of bipolar disorder.
Oxidative phosphorylation and glycolysis
A study by Naydenov et al. (2007), in which they obtained lymphocytes from bipolar patients and healthy controls and examined differences in the pattern of expression of ETC genes in response to energy stress, provides insight into the possible nature of mitochondria dysregulation in bipolar disorder. This study showed no difference between bipolar patients and healthy controls in the expression of ETC genes under normal glucose concentrations. When cultured lymphocytes were exposed to glucose deprivation, however, bipolar patients displayed reduced expression of ETC genes while healthy controls uniformly increased ETC gene expression. This pattern suggests an inability of mitochondria in bipolar patients to adapt to energy stress.
The impaired response to energy stress observed in bipolar disorder described above is associated with several measurable effects of mitochondrial dysfunction including lower intracellular pH levels, increased lactate levels, and a decrease in overall energy production (Dager et al., 2004; Stork and Renshaw, 2005). Studies utilizing MRS showed that compared to normal controls, bipolar patients not taking medication had increased cerebral lactate levels, specifically in the gray matter (Dager et al., 2004). Moreover, lower pH levels have been reported in the basal ganglia region and whole brain of euthymic bipolar subjects in comparison to individuals without bipolar disorder (Hamakawa et al., 2004).
Under energy stress, mitochondria usually rely on glycolysis for a quick source of ATP energy. During this anaerobic cellular respiration process, nicotine adenine dinucleotide (NADH) is converted to NAD, thereby reducing pyruvate to lactate and causing a subsequent decrease in intracellular pH levels. While glycolysis is a readily available source of energy, it only manufactures 2 ATP per glucose molecule in contrast to the 36 ATP produced through oxidation phosphorylation (Ng et al., 2008). In bipolar disorder, MRS studies have documented increased lactate (Dager et al., 2004; Kato et al., 1998) and lowered pH levels (Hamakawa et al., 2004; Kato et al., 1998) suggesting that neurons are constantly relying on the largely inefficient process of glycolysis to produce energy. Overall, these findings provide evidence that, compared to normal controls, the total energy output of mitochondria in bipolar disorder is inhibited.
Brain energy metabolism in bipolar disorder
Multiple reports using MRS have also suggested that bipolar disorder is characterized by abnormalities of brain energy metabolism, consistent with mitochondrial dysfunction. Proton MRS (1H-MRS) studies show decreased cerebral N-acetyl-aspartate (NAA) in bipolar subjects compared to normal controls (Bertolino et al., 2003; Deicken et al., 2003). Decreased levels of NAA, a marker of neuronal viability, are consistent with impaired mitochondrial energy production (Clark, 1998) as NAA is of mitochondrial origin: NAA is synthesized in mitochondria by the membrane-bound enzyme L-aspartate N-acetyltransferase, a catalyst that is found only in the brain (Patel and Clark, 1979).
Phosphorus MRS (31P-MRS) studies allow measurements of high-energy compounds such as ATP and PCr – a reservoir for the generation of ATP that has been found to be altered in bipolar patients (Kato et al., 1994, 1995). During periods of acute neuronal activity, as ATP is utilized and depleted, PCr is rapidly broken down in order to maintain the overall concentration of ATP (Erecinska and Silver, 1989). Long-term abnormalities in PCr levels generally reflect much larger alterations in cellular metabolism and, in particular, an insufficient supply of the ATP needed for normal cellular function (Rothman, 1994). Therefore, continually decreased levels of PCr are suggestive of hypometabolism, possibly due to mitochondrial dysfunction (Modica-Napolitano and Renshaw, 2004). Persistently low brain PCr values have also been found in patients with a variety of mitochondrial disorders (e.g. mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), Leigh’s disease, progressive external ophthalmoplegia (PEO), and Leber’s hereditary optic neuropathy) (Barbiroli et al., 1993; Eleff et al., 1990).
Indirectly, abnormalities of lipid metabolism, an energy-intensive process (Purdon and Rapoport, 1998), are also supportive of the mitochondrial dysfunction and associated bioenergetic deficits in bipolar disorder. Multiple studies have suggested that total choline levels are elevated in bipolar disorder (Hamakawa et al., 1998; Lafer et al., 1994; Moore et al., 2000). Phosphomonoester (PME) levels (which primarily include the lipid membrane precursors phosphocholine and phosphoethanolamine) are also altered in bipolar disorder. Euthymic bipolar patients show decreased PME levels compared with healthy volunteers (Yildiz et al., 2001). Both series of findings suggest impaired lipid membrane metabolism in bipolar disorder. This is important, as lipid membrane turnover represents 10–15% of total energetic requirements in the cell (Purdon and Rapoport, 1998).
Mitochondrial gene expression in bipolar disorder
Multiple mitochondrial genes have been found to be upregulated or downregulated in bipolar disorder compared to normal controls (Benes et al., 2006; Munakata et al., 2004; Naydenov et al., 2007; Sun et al., 2006; Wang, 2007). Mitochondrial DNA (mtDNA) contains 37 genes, 13 encoding for subunits of the respiratory chain complexes I, III, IV and V, 22 encoding for transfer RNAs, and two for ribosomal RNAs (Taylor and Turnbull, 2005). MtDNA differs somewhat from autosomal (nuclear) DNA: it is not protected by any repair mechanism or histones, does not undergo any recombination during meiosis, and is maternally inherited. Notably, polymorphisms are estimated to occur up to 10 times more frequently than in autosomal DNA, probably owing to closeness to free radical production and absence of an effective DNA repair mechanism (Taylor and Turnbull, 2005).
Because of the compact nature of mtDNA, it has been demonstrated that genotyping a small number of single-nucleotide polymorphisms can capture nearly all common variation in the mitochondrial genome. Specifically, Saxena and colleagues reported a set of SNPs that efficiently ‘tag’ common mitochondrial variation (Saxena et al., 2006). In light of the evidence presented above, such variations would have a high prior probability of association with differential treatment response.
Mitochondrial variation is also of particular interest in light of the recent association between calcium channel variation and bipolar disorder (Ferreira et al., 2008; Sklar et al., 2008). Specifically, mitochondria and endoplasmic reticulum (ER) function in concert to sequester Ca2+ ions after their temporary diffusion in subcellular regions. A massive movement of positively charged Ca2+ into the mitochondrion could exceed its capacity to export cations, impairing ATP synthesis and promoting apoptosis (Bernardi et al., 1998). Of note, animal, in vitro and mRNA studies demonstrate that chronic lithium treatment, as well as therapeutic doses of valproic acid and electroconvulsive stimulus, increase the expression of Bcl-2 (Chang et al., 2009; Chen et al., 1999). In turn, one of the main effects of the anti-apoptotic protein Bcl-2 is increasing the stability of the mitochondrial permeability transition pore (MPTP) (Weeber et al., 2002).
Bcl-2, BDNF, neuroprotection and mitochondria as pathophysiology and targets of treatments
Mitochondria play important roles in the regulation of intracellular calcium (Ca2+), and in the general process of synaptic plasticity (Yang et al., 2003). Excessive opening of N-methyl-d-aspartate (NMDA) glutamate receptors can trigger large influxes of Ca2+, which in turn can overwhelm the ability of mitochondria to export protons and cations (Nicholls and Ward, 2000) and can lead to drastic reduction in ATP synthesis (Weeber et al., 2002); at the extreme, this can lead to initiation of apoptosis (Bernardi et al., 1998). Activation of mitochondrial apoptotic mechanisms may lead to selective destruction of synapses (Culmsee and Mattson, 2005). This may be related to neuroplasticity and synaptic pruning in normal conditions, but is also involved with extensive loss of synapses in pathological conditions associated with mitochondrial dysfunction (Mattson and Liu, 2003).
In bipolar disorder, patients with the val66met polymorphism of the BDNF gene (which may be associated with rapid cycling (Müller et al., 2006)) also have low levels of PCr + Cr in the dorsolateral prefrontal cortex (suggestive of low bioenergetic metabolism (Frey et al., 2007b)). Rats treated with therapeutic doses of lithium and valproic acid have increased levels of Bcl-2 (Manji et al., 2000). Bcl-2 and other neuroprotective agents from its family (Bcl-xl) exert their anti-apoptotic and neuroprotective activity by increasing the ability of mitochondria to resist the toxic effect of elevated intracellular Ca2+ (Murphy et al., 1996).
The presence of mitochondrial dysfunction in bipolar disorder and the link between current effective treatments in bipolar disorder (lithium, valproic acid), major neuroprotective factors (Bcl-2, BDNF and GSK-3) and their impact on mitochondrial activity may justify enhancing mitochondrial function as a putative target in the search of future mood stabilizers.
Candidate mitochondrial modulators
Several readily available dietary supplements (or nutraceuticals) have been tested as potential treatments in bipolar disorder (for a review see Sarris et al., 2011). Among them, a subset of substances has the potential to improve mitochondrial function and brain energy metabolism; those are grouped here under the term ‘mitochondrial modulators’. Candidates include N-acetyl-cysteine (NAC), acetyl-L-carnitine (ALCAR), S-adenosylmethionine (SAMe), coenzyme Q10 (CoQ10), alpha-lipoic acid (ALA), creatine monohydrate (CM), and melatonin. As pointed out by Hagen and colleagues, increased mitochondrial metabolism can result in decreased antioxidant status – and the decreased antioxidant status may need to be offset by providing an additional antioxidant such as ALA (Hagen et al., 2002a) – alternatively, NAC may provide the sufficient antioxidant properties (by increasing glutathione) without ALA. Furthermore, allostatic load in bipolar disorder has been reported to be increased, as evidenced by high levels of ROS (Kapczinski et al., 2008) and MMs could prevent ROS damage.
N-acetyl-cysteine (NAC)
NAC, by supplying cysteine, increases synthesis of the free radical scavenger glutathione (GSH) which, in turn, reduces oxidative stress (Atkuri et al., 2007). Glutathione (GSH), a tripeptide composed of glutamate, cysteine and glycine, is the most abundant thiol antioxidant. Glutamate-cysteine ligase (GCL) is the rate-limiting enzyme for GSH production – GCL ligates glutamate with cysteine to form gamma-glutamyl-cysteine which is then combined with glycine and catalyzed by GSH synthase to form GSH (Cui et al., 2007). As an antioxidant, GSH reacts with hydrogen peroxide H2O2 to form H2O with the byproduct of GSH disulfide. Also, GSH can conjugate with oxidized products, catalyzed by glutathione-S-transferase (GST) to further reduce oxidative stress.
Lithium and valproate have well-known neuroprotective effects that may be mediated by increasing GSH and GCL levels. In cultured rat cerebral cortical cell cultures, the neuroprotective effects of lithium and valproate against oxidative damage from H2O2 is abolished by depletion of GSH (Cui et al., 2007). Furthermore, one mechanism of neuroprotection of lithium may be through GSH, since lithium increases the gene expression of GST isoenzymes (Shao et al., 2008a). NAC can increase GSH further (and potentially synergistically with lithium or valproate) by driving synthesis of glutathione to the right with increased cysteine availability.
Oral NAC prevents glutathione depletion in the brain by increasing cysteine and synthesis of glutathione systemically (Berk et al., 2008a/b). Furthermore, NAC has been found to be neuroprotective and can prevent oxidative damage in complex 1 in the mitochondrial electron transport chain (Mayer and Noble, 1994; Nicoletti et al., 2005). Clinically, NAC augmentation reduced negative symptoms and improved auditory processing in schizophrenia (Berk et al., 2008a/b), improved pathological gambling in a pilot study (Grant et al., 2007), and is a prime candidate to improve mitochondrial function, and perhaps the long-term course, in bipolar disorder.
In a preliminary, double-blind study of bipolar patients (n = 75) who had experienced a mood episode in the past 6 months and who had been stable for at least 1 month, NAC 2 grams per day or placebo was added to ongoing medications (Berk et al., 2008a/b). At baseline, half the patients were euthymic, about a third were depressed, and about 15% had euphoric or dysphoric mania. Measures of depression, quality of life, and functioning were all better with NAC compared to placebo beginning at week 12 and continuing until week 24; no differences were found in manic or hypomanic symptoms, but these symptoms were minimal at baseline and during the study. The delayed effect of NAC (observed first at week 12) may be a result of the study design, as most patients were euthymic at baseline. Improvements in mood that occurred during the trial stopped after discontinuation of NAC.
Acetyl-L-carnitine (ALCAR)
L-Carnitine, composed of lysine and methionine, can be endogenously generated or obtained through diet (Hoppel, 2003). Carnitines transport fatty acids into mitochondria for beta-oxidation and energy generation, and scavenge ROS (Al-Majed et al., 2006; Rebouche, 2004). These fatty acids enter mitochondria as acyl-carnitines, and when oxidized, release energy and form acyl-coenzyme A, which then enters the citric acid cycle (Hoppel, 2003). ALCAR, absorbed better than L-carnitine and more able to cross the blood–brain barrier than L-carnitine (Ames and Liu, 2004a), has neuroprotective and anti-apoptotic properties (Al-Majed et al., 2006; Virmani et al., 2005) and has been shown to block glutamate-induced over-expression of glutamic acid decarboxylase GAD67 (Hao et al., 2004), the main enzyme that converts glutamate to gamma-amino-butyric acid (GABA). ALCAR appears to reverse age-related degeneration in animal models (Aliev et al., 2009; Ames and Liu, 2004a, 2004b; Hagen et al., 2002a; Liu et al., 2002b) and may slow down or reverse age-related cognitive and motoric decline in rats, as well as reverse diminished reactivity to the environment (Hagen et al., 2002a) – effects thought to result from enhanced mitochondrial functioning. Notably, adult and aged rats administered ALCAR both show an increase in brain levels of ATP and PCr as measured by 1H nuclear magnetic resonance spectroscopy (NMR) (Aureli et al., 1990). However, evidence that ALCAR can decrease oxidative stress is mixed (Ames and Liu, 2004a), suggesting that it may be more clinically effective when administered with a potent antioxidant such as NAC or ALA (see below).
Clinically, ALCAR has shown benefit in clinical trials for a number of neuropsychiatric disorders including dementia (Bonavita, 1986; Passeri et al., 1990; Pettegrew et al., 1995; Salvioli and Neri, 1994; Sano et al., 1992; Spagnoli et al., 1991), geriatric depression (Bella et al., 1990; Fulgente et al., 1990; Garzya et al., 1990; Gecele et al., 1991; Nasca et al., 1989; Pettegrew et al., 2002; Tempesta et al., 1987; Villardita et al., 1983), dysthymia (Zanardi and Smeraldi, 2006), hyperactivity associated with fragile X syndrome (Torrioli et al., 1999, 2008), ADHD, inattentive type (Arnold et al., 2007), degenerative cerebellar ataxia (Sorbi et al., 2000), minimal hepatic encephalopathy (Malaguarnera et al., 2008), and hepatic coma secondary to cirrhosis (Malaguarnera et al., 2006), and has demonstrated benefit in disorders thought to be associated with deficits in neuronal mitochondrial energy production such as diabetic peripheral neuropathy (De Grandis and Minardi, 2002; Sima et al., 2005) and HIV-associated antiretroviral toxic neuropathy (Hart et al., 2004; Youle and Osio, 2007). One study investigating the weight-reducing effects of ALCAR in bipolar patients who had gained weight while taking valproate found no difference in weight between those taking ALCAR with diet compared to diet alone (Elmslie et al., 2006). Interestingly, valproate-induced hepatotoxicity may be mediated by inhibition of mitochondrial fatty acid beta-oxidation and depletion of carnitine, suggesting that carnitine supplementation, perhaps through administration of ALCAR, may either prevent or reverse valproate-induced hepatotoxicity (Silva et al., 2008).
Importantly, ALCAR has demonstrated efficacy in a variety of depressive spectrum disorders in several placebo-controlled studies (Bella et al., 1990; Fulgente et al., 1990; Garzya et al., 1990; Gecele et al., 1991; Nasca et al., 1989; Pettegrew et al., 2002; Tempesta et al., 1987; Villardita et al., 1983; Zanardi and Smeraldi, 2006). In addition, a preliminary study in depressed geriatric patients demonstrated improvement in depression symptoms that were associated with an increase in PCr as measured by 31P-MRS after monotherapy treatment with ALCAR (Pettegrew et al., 2002). However, no clinical trials, to our knowledge, have yet examined the effect of ALCAR supplementation in individuals with bipolar disorder.
S-Adenosylmethionine (SAMe)
SAMe, formed from methionine and ATP, is a naturally occurring biological component of all living cells, and plays a critical role in cellular metabolism as a major source of methyl groups for key biochemical reactions (Bottiglieri, 1996; Bottiglieri et al., 2000). Specifically as a methyl donor, SAMe is a precursor molecule for pathways catalyzed by methyltransferase enzymes, such as methylation, transulfuration and aminopropylation.
Methylation via SAMe is a particularly important process in the central nervous system. SAMe is integral in the degradation and repair of proteins with aberrant levels of
SAMe is also a precursor molecule for GSH production through transulfuration. S-Adenosyl-homocysteine (SAH), formed when SAMe donates its methyl group, is rapidly metabolized to homocysteine. Homocysteine, when paired with several B vitamin cofactors, including pyridoxal phosphate (vitamin B-6), is converted into cystathionine and eventually, through transulfuration, converted to GSH. As discussed earlier in the section on NAC, GSH plays an essential role in reducing oxidative stress. Homocysteine can also regenerate SAMe when a sufficient concentration of B vitamins triggers its remethylation.
Clinically, SAMe evidence supports the antidepressant effects for individuals with depressive disorders, with fewer side effects than standard medications such as imipramine, chlorimipramine, nomifensive and minaprine (Bottiglieri, 1996; Bottiglieri et al., 2000). The results of an open label trial of SAMe treatment in depressed inpatients supported the clinical value of SAMe supplementation such that patients experienced remission or improvement of their symptoms with minimal side effects after a 2-week period (Lipinski et al., 1984). More recent research has investigated the efficacy and tolerability of SAMe when combined with other traditional antidepressant medication. When SAMe was added to selective serotonin reuptake inhibitors (SSRI) antidepressants or venlafaxine, individuals with unremitted depression showed significant improvements, including a decrease in both depression-severity and clinical global impressions severity scores as assessed by the HAM-D and CGI scales, respectively (Alpert et al., 2004). In a recent randomized controlled trial, SAMe augmentation of antidepressants was more efficacious than placebo augmentation in unipolar depressed subjects not responding to a course of selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs) (Papakostas et al., 2010). While evidence for efficacy exists when SAMe is combined with imipramine (Berlanga et al., 1992), the results for SAMe augmentation with older antidepressants, including phenelzine, mianserin and maprotiline (Alvarez et al., 1987) showed inconsistent results. Of note, SAMe may also carry a risk of manic switch in bipolar disorder. In an earlier study, nine of 11 bipolar patients treated with SAMe switched into elevated mood state (hypomania, mania and euphoria) while the other two did not respond (Carney et al., 1989). To the best of our knowledge, no systematic randomized controlled studies of SAMe for bipolar disorder have been conducted to date.
Coenzyme Q10 (CoQ10)
Coenzyme Q10 (CoQ10) consists of polyisopropyl units (usually 9 or 10) attached to a benzoquinone ring that can alternate between free radical ubisemiquinone (*QH), and fully reduced ubiquinol (QH2) (Kwong et al., 2002). The ability to assume multiple redox states allows for CoQ10 to play several functional roles in the cells, not all of which are well understood. CoQ10 acts as a lipid-soluble antioxidant that can prevent cell damage by neutralizing free radicals directly or by reducing an alpha-tocopheroxyl radical to alpha-tocopherol, an absorbable form of vitamin E. Another important function of CoQ10, present in high concentrations within the inner mitochondrial layer, is its ability to generate an electrochemical gradient along the transmembrane of mitochondria. The quinine head of CoQ transfers electrons in the mitochondrial electron transport chain from complexes I and II to complex III while releasing protons into the intermembrane.
While CoQ10 naturally decreases with aging in various tissues, abnormal levels of CoQ10 are also associated with conditions such as hypertension, diabetes, cardiovascular disease, and neurodegenerative disorders such as Parkinson’s disease.
Plasma and mitochondrial brain levels of CoQ10 in individuals with Parkinson’s disease were significantly lower than non-parkinsonian controls of the same gender and age (Shults and Haas, 2005). While the total level of plasma CoQ10 was reduced in subjects with Parkinson’s disease, the percentage of oxidized plasma CoQ10 (ubiquinone-10) was increased (Sohmiya et al., 2004). Because CoQH2–10 (e.g. ubiquinol-10) is oxidized to CoQ10 with early plasma oxidation, high levels of CoQ10 may serve as a biomarker of oxidative stress in the mitochondria. A significant correlation was also found between the level of CoQ10 and the activity levels of complex I and II/III. Impaired activity of complex I, II and III was observed in platelet mitochondria of patients with early, untreated Parkinson’s disease (Shults et al., 1997).
Evidence for mitochodrial dysfunction in bipolar (BP) disorder.
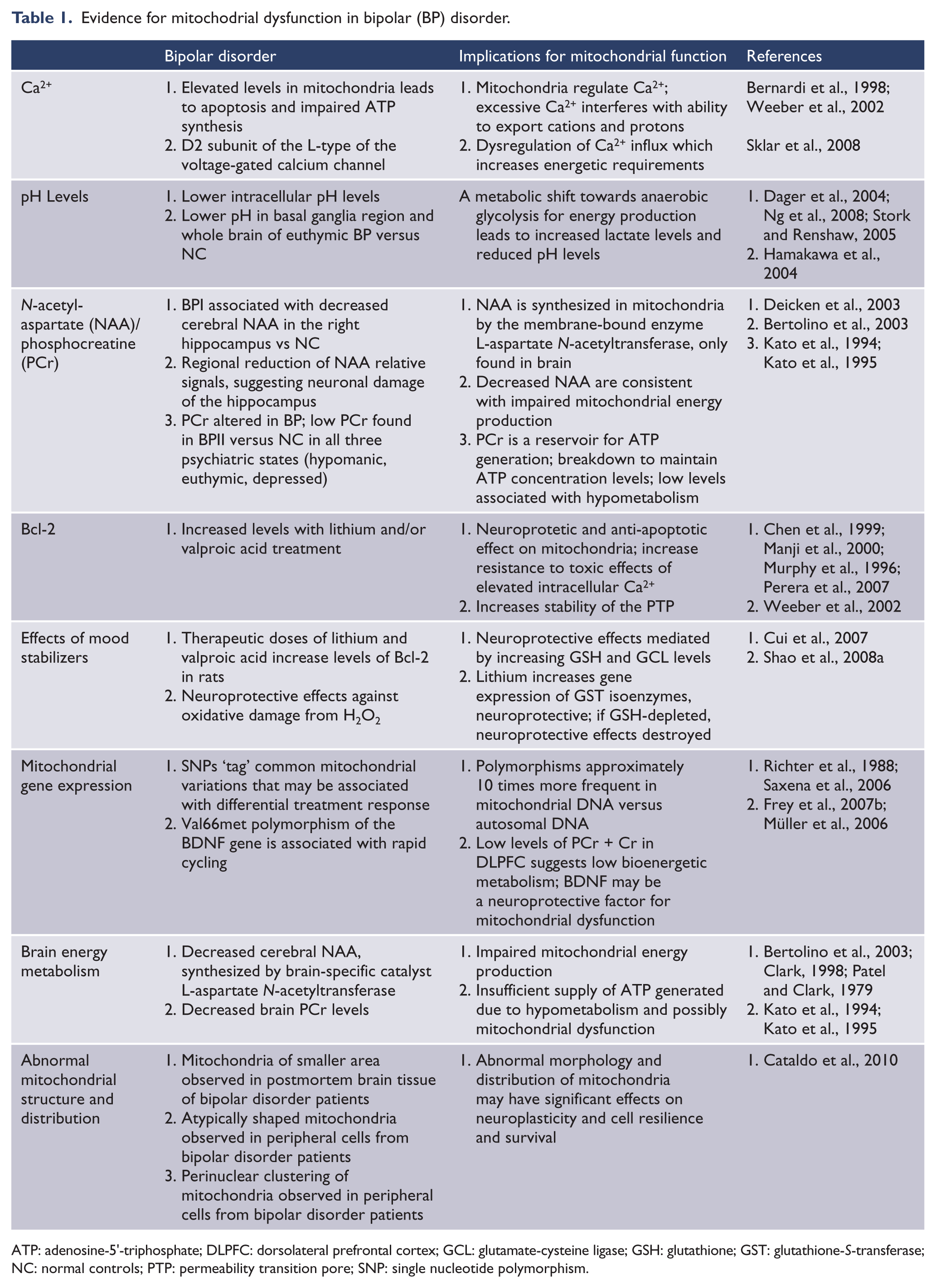
ATP: adenosine-5′-triphosphate; DLPFC: dorsolateral prefrontal cortex; GCL: glutamate-cysteine ligase; GSH: glutathione; GST: glutathione-S-transferase; NC: normal controls; PTP: permeability transition pore; SNP: single nucleotide polymorphism.
Potential mitochondrial modulators and their effects on mitochondria and oxidative stress.
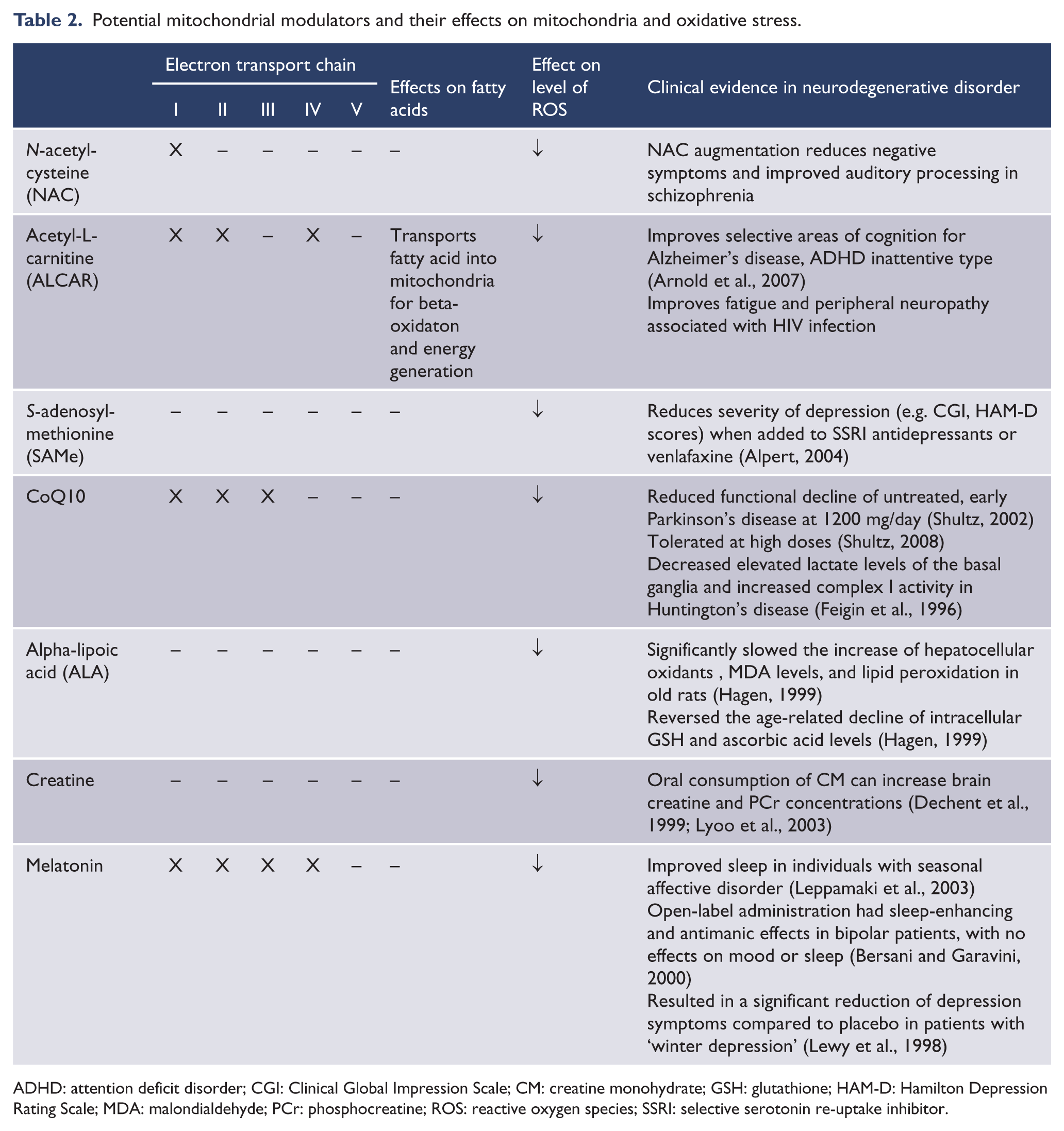
ADHD: attention deficit disorder; CGI: Clinical Global Impression Scale; CM: creatine monohydrate; GSH: glutathione; HAM-D: Hamilton Depression Rating Scale; MDA: malondialdehyde; PCr: phosphocreatine; ROS: reactive oxygen species; SSRI: selective serotonin re-uptake inhibitor.
Evidence of CoQ10’s role in oxidative stress suggests that oral administration of coenzyme Q10 has the potential to be part of the treatment of such neurodegenerative diseases. For example, individuals with Huntington’s disease showed a decrease in elevated lactate levels in the basal ganglia, as well as an increase in complex I activity after treatment with CoQ10 (Feigin et al., 1996). Patients with early, untreated Parkinson’s disease showed a slower decline in functioning when CoQ10 was given at a dose of 1200 mg/day (Shults et al., 2002), a dose that has been found to have a good safety profile (Hidaka et al., 2008). A more recent study investigating a higher dosage of CoQ10 in conjunction with vitamin E suggested that CoQ10 is safe and tolerated at doses up to 3000 mg/day for 2 weeks, but should not exceed 2400 mg/day since any dosage beyond this amount does not increase plasma levels of alpha-tocopherol (Shults et al., 2004). Other evidence found that while CoQ10 supplementation increased plasma levels of CoQ10 in patients with Parkinson’s disease, it had no effect on Parkinsonian symptoms after a 3-month period (Storch et al., 2007). Similarly, in an open label trial of 200 mg of daily CoQ10 supplementation, idiopathic parkinsonian patients demonstrated no significant clinical effect in a 3-month period (Strijks et al., 1997).
Clinical trials of CoQ10 in other health conditions also indicate evidence of clinical improvement with CoQ10 supplementation. In a recent meta-analysis of 12 clinical trials of the efficacy of CoQ10 supplementation in hypertensive patients, it was suggested that CoQ10 may lower both systolic blood pressure and diastolic blood pressure (Rosenfeldt et al., 2007). A 3-month supplementation of CoQ10 in patients with migraine showed a reduction in the frequency and disability of headaches (Hershey et al., 2007). Similarly, individuals randomized to CoQ10 supplementation (300 mg/day) in a double-blind, randomized, placebo-controlled trial showed improvement in symptoms of migraine including reduced attack-frequency, reduced nausea and vomiting associated with migraine, and fewer headache days over a 4-month period (Sandor et al., 2005). Such evidence suggests that the level of CoQ10 may be helpful in both understanding and treating disorders related to dysfunctional energy metabolism, particularly mood disorders. To our knowledge, no clinical trials have been completed that have investigated the clinical value of CoQ10 for individuals with major depressive disorder, bipolar disorder, or anxiety disorders.
Alpha-lipoic acid (ALA)
Alpha-lipoic acid (ALA) is a naturally occurring mitochondrial coenzyme for pyruvate and alpha-ketoglutarate dehydrogenase. It is a potent antioxidant (Packer et al., 1995) and carries out an essential role in mitochondrial energy production by recruiting glucose transporters to the cell membrane, thereby increasing the cellular uptake of glucose for ATP synthesis (Estrada et al., 1996).
ALA can be obtained through diet in foods such as spinach, yeast and red meats, and has been shown to have antioxidant properties when administered as a dietary supplement to increase the unbound fraction of the compound. Both in vitro and in vivo studies of ALA supplementation have demonstrated potent antioxidant effects (Packer et al., 1995). For example, one study found that ALA supplementation slowed the increase of hepatocellular oxidants, malondialdehyde (MDA) levels and lipid peroxidation in old rats, and reversed the age-related decline of intracellular GSH and ascorbic acid levels (Hagen et al., 1999).
In addition to improving cellular antioxidant status, ALA has been shown to reverse metabolic deficits, oxidative stress, and damage associated with aging when administered as a dietary supplement (Hagen et al., 2002a). Moreover, ALA has been shown to effectively reduce oxidative stress and ameliorate energy-related metabolic impairments, specifically in type II diabetes, liver disease or age-related oxidative damage (Hagen et al., 2002a). Specifically, high-dose ALA administered intravenously to diabetic and control rats induced a rapid reduction of blood glucose levels and acetyl CoA content in the liver (Rudich et al., 1999), and oral doses of ALA lowered elevated hepatic and cerebral malondialdehyde levels of older rats (Hagen et al., 2002b).
ALA supplementation has also demonstrated neuroprotective effects against peripheral oxidative stress and damage from
ALA has been investigated as a potential treatment in a number of diseases thought to be associated with abnormal mitochondrial functioning and impaired energy utilization, including type II diabetes (Jacob et al., 1999) and diabetic polyneuropathy (Ametov et al., 2003; Hahm et al., 2004; Negrisanu et al., 1999; Reljanovic et al., 1999; Ruhnau et al., 1999; Tankova et al., 2004; Ziegler et al., 1995, 1999, 2006). It has also been studied in the treatment of alcohol-related liver disease (Marshall et al., 1982), burning mouth syndrome (Femiano and Scully, 2002), diabetes-related cardiac autonomic neuropathy (Ziegler et al., 1997), HIV-associated cognitive impairment (Anonymous, 1998), multiple sclerosis (Yadav et al., 2005), migraine (Magis et al., 2007), peripheral arterial disease (Vincent et al., 2007) and Alzheimer’s disease (Hager et al., 2007).
Although the specific mechanism by which ALA improves mitochondrial function has not been identified, several have been discussed in the literature including: (1) increased recycling of endogenous antioxidants including CoQ10, vitamins C and E, and GSH (Liu, 2008); (2) reduction to the highly potent antioxidant dihydrolipoic acid which reacts with superoxide radicals, hypochlorous acid, peroxyl radicals, and singlet oxygen (Liu et al., 2002a); (3) chelation of heavy metals such as iron and copper (Liu, 2008); (4) stimulation of glucose uptake into cells leading to enhanced energy metabolism (Estrada et al., 1996); (5) anti-apoptotic and neuroprotective effects secondary to prevention of glutamate-induced increases in intracellular calcium (Liu, 2008; Liu et al., 2002a); and (6) stimulation of mitochondrial biogenesis through upregulation of mitochondrial transcription factors such as peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) (Liu, 2008).
Creatine monohydrate (CM)
Creatine is a non-essential dietary element that is found in meat and fish and is synthesized endogenously by the liver and kidneys (Juhn and Tarnopolsky, 1998; Terjung et al., 2000). Exogenous creatine is widely available as a dietary supplement in the form of creatine monohydrate (CM).
Creatine is the precursor of PCr, which plays an integral role in brain and muscle energy metabolism. The primary role of PCr is to function as a reservoir of inorganic phosphate to be drawn upon to produce ATP by the catalyzing agent creatine kinase (Ames, 2000). During periods of transient high-energy demands, PCr is rapidly converted to creatine in order to donate a high-energy phosphate to adenosine diphosphate (ADP) to maintain the overall intracellular concentration of ATP (Erecinska and Silver, 1989; Sauter and Rudin, 1993). While short-term decreases in PCr may reflect typical physiologic responses to acute muscular or neuronal activity, long-term abnormalities in PCr generally reflect an insufficient metabolic supply of ATP needed for normal cellular function, possibly due to underlying mitochondrial dysfunction, leading to deficits in oxidative phosphorylation (Rothman, 1994). This concept is reflected in studies of patients with mitochondrial cytopathies, in which reduced brain PCr concentrations have been observed (Barbiroli et al., 1993). Decreased PCr concentrations have also been identified in patients with bipolar disorder (Stork and Renshaw, 2005). Notably, several studies have shown that oral consumption of CM can increase brain creatine and PCr concentrations (Dechent et al., 1999; Lyoo et al., 2003), suggesting that CM supplementation may mitigate bioenergetic abnormalities by providing additional substrate for the production of ATP through the creatine kinase reaction.
In addition to enhancing energy production, creatine has been shown to have direct and indirect antioxidant properties (Lawler et al., 2002; Tarnopolsky, 2008; Tarnopolsky and Beal, 2001). For example, creatine has been shown to reduce markers of oxidative stress in animal models of Huntington’s disease (Andreassen et al., 2001) and amyotrophic lateral sclerosis (ALS) (Klivenyi et al., 1999). Moreover, creatine supplementation was found to have indirect antioxidant properties in an animal model of traumatic brain injury that were associated with reduced apoptosis, prevention of opening of the MPTP, and better maintenance of the mitochondrial membrane potential (Lawler et al., 2002; Sullivan et al., 2000).
Several clinical trials with CM in the treatment of mitochondrial cytopathies (Tarnopolsky et al., 1997), neuromuscular disorders (Tarnopolsky et al., 2004; Walter et al., 2000) and neurodegenerative disorders such as Parkinson’s disease (Bender et al., 2006; NINDS NET-PD Investigators, 2006), Huntington’s disease (Tabrizi et al., 2005; Verbessem et al., 2003) and ALS (Groeneveld et al., 2003; Rosenfeld et al., 2008) have been performed with mixed results. CM supplementation demonstrated benefit in one small, open-label study in patients with post-traumatic stress disorder (PTSD) (Amital et al., 2006a) and was associated with improvement in depression symptoms in a patient with comorbid depression and fibromyalgia (Amital et al., 2006b). In contrast, there was no evidence of efficacy following CM treatment in a small controlled trial in schizophrenia (Kaptsan et al., 2007). A small open-label study suggested a beneficial effect of CM augmentation in patients with treatment-resistant depression (Roitman et al., 2007). Interestingly, this study enrolled two bipolar depressed patients – both of whom developed hypomania/mania after CM treatment. All in all, preclinical and clinical evidence to date encourages further exploration of the potential benefit of CM supplementation in disorders associated with mitochondrial dysfunction, including bipolar disorder.
Melatonin
Melatonin was long conceived as a hormone secreted in a tightly regulated circadian rhythm by the pineal gland, which served a primary role in reproduction. However, the recent recognition that melatonin is synthesized in many organs and tissues in the body, including the brain, has sparked exploration into its potential role in a variety of physiological processes.
Growing evidence suggests that melatonin has potent antioxidant properties that result from direct free radical scavenging properties as well as effects on mitochondrial physiology. Several studies have demonstrated that melatonin and several of its metabolites are major scavengers of both oxygen- and nitrogen-based ROS (Korkmaz et al., 2009; Lopez-Burillo et al., 2003; Reiter et al., 2002a, 2002b, 2003), and melatonin has been shown to stimulate the production of GSH (Albarran et al., 2001; Winiarska et al., 2006). Interestingly, melatonin has also been shown to have direct genomic effects. For example, melatonin increases the mRNA expression of genes involved in antioxidant production such as glutathione peroxidase and superoxide dismutase (SOD) under physiological conditions and during elevated oxidative stress (Acuna-Castroviejo et al., 2007; Rodriguez et al., 2004; Tan et al., 1998).
In addition to its antioxidant effects, evidence suggests that melatonin may have a direct benefit on mitochondrial functioning. Both in vitro and in vivo animal studies have demonstrated multiple mitochondrial enhancing effects following melatonin administration, including increased oxidative phosphorylation through activation of ETC complexes, increased mitochondrial membrane fluidity, and closing of the MPTP (Acuna-Castroviejo et al., 2001, 2007; Garcia et al., 1997; Leon et al., 2004, 2005). Moreover, melatonin has direct effects on mitochondrial genome expression. For example, studies have shown that melatonin administration prevents oxidative degradation of mtDNA and reduction of mtDNA transcripts in several tissues including brain (Acuna-Castroviejo et al., 2005) and increases expression of mtDNA-coded polypeptide subunits I, II and III of ETC complex IV (Acuna-Castroviejo et al., 2003).
Recent exploration into the potential clinical utility of melatonin, primarily in the treatment of neurodegenerative disorders hypothesized to result, in part, from mitochondrial dysfunction, has provided some encouraging results. Melatonin prevented neurodegenerative changes in several experimental models of Alzheimer’s disease (Lahiri et al., 2004; Pappolla et al., 1998; Reiter et al., 1999) and Huntington’s disease (Reiter et al., 1999). Furthermore, preliminary studies in Alzheimer’s disease patients have demonstrated a slowing of cognitive impairment and a reduction in sundowning following melatonin treatment (Cardinali et al., 2002). In addition, melatonin has demonstrated neuroprotective properties in animal models of Parkinson’s disease hypothesized to result from ROS scavenging, enhancement of ETC activity and increased mitochondrial membrane potential (Acuna-Castroviejo et al., 2007; Reiter et al., 1999).
Several small studies have evaluated the effect of melatonin administration in mood disorders with mixed results. A controlled study of melatonin in seasonal affective disorder demonstrated improvement in sleep and vitality, but no reduction in atypical depression symptoms (Leppamaki et al., 2003). Patients with ‘winter depression’ demonstrated significantly reduced depression symptoms following melatonin treatment compared to placebo (Lewy et al., 1998). In contrast, melatonin was associated with worsening dysphoria and reduced sleep and weight in a crossover study in patients with severe depression (Carman et al., 1976). With regard to bipolar disorder, open-label melatonin had no significant effects on mood or sleep in a small study of rapid-cycling bipolar disorder (Leibenluft et al., 1997) but was found to have sleep-enhancing and antimanic effects in a small open-label study in bipolar mania (Bersani and Garavini, 2000).
Interestingly, agomelatine, a potent agonist of melatonin MT1 and MT2 receptors, has demonstrated preliminary evidence of efficacy in bipolar depression (Calabrese et al., 2007). However, the antioxidant and mitochondrial-enhancing properties of melatonin are speculated to result primarily from direct free radical scavenging and actions linked to cytosolic proteins rather than receptor-mediated mechanisms (Acuna-Castroviejo et al., 2007). Nevertheless, further exploration into the potential for melatonin-receptor modulators to enhance mitochondrial functioning is warranted.
Rationale for considering combinations of mitochondrial modulators
While convergent data strongly support the hypothesis that mitochondrial dysfunction occurs in bipolar disorder, multiple abnormalities along the mitochondrial ETC can occur, making it improbable that, for any given patient, the correct abnormality can be practically identified with currently available technology. In addition, while compounds such as ALCAR, SAMe and CM may increase energy production, this increase may also cause an increase in the production of ROS (Hagen et al., 2002a). Mitochondrial DNA lack histones and are therefore extremely susceptible to damage from ROS. This feature sets up a vicious cycle in which increased ROS generation leads to further damage to mitochondrial DNA, proteins and lipids, which can further exacerbate defects in mitochondrial energy production.
Therefore, effective mitochondrial enhancement strategies may require several agents that both modulate multiple mitochondrial targets and increase mitochondrial metabolic activity without a concomitant increase in oxidative stress – a strategy that has been proposed previously in the treatment of mitochondrial disorders (Rodriguez et al., 2007; Tarnopolsky, 2008).
This ‘mitochondrial cocktail’ could include several of the agents described above. For example, whereas NAC targets complex I in the ETC and reduces oxidative stress, ALCAR targets complexes I, II and IV, as well as enhancing oxidation of fatty acids. Therefore, we reason that the combination treatment of NAC with ALCAR is more likely to modulate multiple mitochondrial targets while mitigating the resulting oxidative stress from ALCAR-induced mitochondrial energy production (Hagen et al., 2002a) by increasing GSH via NAC supplementation. One study found improved cognitive functions and decreased aggression after treatment with a combination of NAC, ALCAR and SAMe in mouse models (Shea, 2007), providing support for this strategy.
Given the enhancing effects of ALCAR on cerebral energy metabolism and the potent antioxidant properties of ALA, we theorize that a combination of ALCAR and ALA might also be effective in increasing mitochondrial metabolic activity without a concomitant increase in oxidative stress – as others have proposed (Hagen et al., 2002a; Soczynska et al., 2008). Animal studies have demonstrated that the combination treatment of ALCAR and ALA improves mitochondrial functioning by increasing metabolism and lowering oxidative stress more than either compound alone (Hagen et al., 2002a; Liu et al., 2002a, 2002b), providing preclinical support for this rationale.
The aim of this review is to support the rationale for rigorous studies of MMs (and combinations) in bipolar disorder. While clinicians could recommend such treatments currently, rigorous data are lacking in most instances to do so. Of note, combinations of MMs are likely to be well tolerated. For all the individual dietary supplements reviewed here, the available literature suggests adequate tolerability and low side-effect burden. While some of these dietary supplements can be costly (and none is reimbursed by insurance), it is unlikely that cost considerations will limit the availability of such treatments, especially after proof of clinical efficacy that will be obtained through rigorous clinical trials.
Summary
While mitochondrial disorders can present in any system, the central nervous system is particularly vulnerable to dysregulations of mitochondrial function since it has high energy demands. Because convergent data implicate subtle mitochondrial dysfunctions as an important component of the pathophysiology of bipolar disorder, it makes sense to consider clinical trials of substances that have the potential to ameliorate those dysfunctions. We reviewed reasonable candidates for modulating mitochondrial function: N-acetyl-cysteine (NAC), acetyl-L-carnitine (ALCAR), S-adenosylmethionine (SAMe), coenzyme Q10 (CoQ10), alpha-lipoic acid (ALA), creatine monohydrate (CM) and melatonin, and postulated that combinations of two or even more of these MMs could be of interest. These MMs should be considered for evaluation in rigorous, well-designed, well-powered clinical trials to improve the long-term course of bipolar disorder.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.