
Abstract
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a rare and serious immunological syndrome that involves a strong activation of cytotoxic T lymphocytes and macrophages. HLH determines a cytokine-mediated tissue injury with a contemporary multi-organ failure and a high fatality rate.
Material and methods
A retrospective study was performed considering the medical records of paediatric patients who underwent a bone marrow aspirate for suspect HLH. The biomarkers evaluated were among those included in the HLH-2004. Lactate dehydrogenase (LD) was also evaluated. Haemophagocytosis was evaluated in bone marrow blood smear slides.
Results
Enrolled were 11 patients included in the HLH group and 8 patients as controls. Haemoglobin and fibrinogen resulted lower in HLH patients than in controls, while blood triglycerides, serum ferritin and LD resulted increased. Blood triglycerides and fibrinogen discriminated HLH cases perfectly, with a sensitivity and specificity of 100%. Ferritin had a sensitivity of 100% and a specificity of 83% (cut off ≥3,721 µg/L) and LD of 73% and of 100% (the cut off ≥1,903 U/L). Haemoglobin was found to have a sensitivity of 75% and a specificity of 100% (cut off ≤ 96 g/L). Total haemophagocytes cell counts were not different between patients and controls. Only the increased number of phagocytized nucleated red blood cells (NRBC) was found to be significantly increased in the patients. Erythrocytes phagocytosis (≥4/1,000 cells) only tended towards significance.
Conclusions
The blood biomarkers showed better diagnostic performance than the morphological evaluation. Among the different cell lineages engulfed by haemophagocytes, the best diagnostic performance was obtained by phagocytosed mature erythrocytes and immature nucleated erythrocytes.
Keywords
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a rare and severe immunological syndrome, characterized by uncontrolled activation of cytotoxic T lymphocytes and macrophages. HLH triggers a cytokine-mediated tissue damage that can lead to multi-organ failure. The key soluble mediators in the immunopathology of HLH are interferon gamma, interleukin 1-beta and interleukin 18.1–3 The main clinical manifestations of HLH are fever, hepatosplenomegaly, coagulopathy, liver damage, pancytopenia, high serum ferritin, reduced or absent cytolytic NK activity. Early diagnosis and treatment are essential for patient survival because HLH can lead to a rapid clinical deterioration with a high fatality rate, if untreated. 4
Despite its severity, HLH is often underdiagnosed. HLH affects both adults and children and can be classified into a primary/familial and a secondary/sporadic form. The primary form usually manifests in early childhood and is caused by high-penetrance homozygous genetic mutations in some of the genes that affect cytolytic functions, lymphocyte survival or inflammatory activation (mainly of CD8 + T cells and NK cells) such as PFR1, UNC13D, STX11, STXBP2, LIST, RAB27A, XIAP, SH2D1A and NLRC4.5,6 The secondary/sporadic forms are linked to acquired factors such as chronic inflammation, autoimmunity, infection or malignancy. 7 In particular, infectious diseases can affect the HLH disease course depending on the trigger. Different infectious agents are responsible for secondary HLH. Among them, some are viruses such as the human immunodeficiency virus (HIV), the hepatitis viruses B and C and Epstein Barr Virus (EBV) and some are bacteria such as Mycobacterium tuberculosis and Pneumocystis jiroveci. 8 Other important causes of secondary HLH, mainly in adults, are viral and bacterial tick-borne infections among which the most important are reported to be caused by the bacteria: Anaplasma phagocytophilum, Ehrlichia chafeensis and Rickettsia rickettsia. 9
Overall, the incidence of HLH primary forms has been estimated to be 1.2 cases per million/year.10–14 These forms account for up to 25% of reported cases and typically affect children under 1 year of age 15 The Swedish national register reports an incidence of Familial HLH of up to 0.15 per 100,000 children/year. 10 Indeed, in neonatal age, the rate of HLH is estimated to be higher, between 1/50,000 and 1/150,000 births. 16 According to a retrospective study conducted in Texas, the cross-sectional prevalence of HLH in children resulted about 1 in 100,000 without gender preference and has been estimated that up to 1 in 3,000 cases admitted to tertiary paediatric hospitals were for HLH.17,18 However, prevalence and incidence may vary in different geographical regions and increase in areas with higher rates of inbreeding. 15
Secondary HLH forms usually affect children over 1 year of age and adults in whom the distribution is usually bimodal, with peaks in younger adults (16–30 years) and older adults (56–70 years). 19 However, incidence and prevalence are not exactly known in adults. 20 A review of 775 cases indicated a mean age of onset of 49 years and a male-to-female ratio of 1.7/1. 21
The first attempt to introduce standardized diagnostic criteria for HLH was made in 1991 by the Histiocyte Society (HLH Study Group). 22 These criteria were modified over time and updated into the HLH-2004 criteria to which three additional parameters were added. The HLH-2004 diagnostic guidelines include at least 5 of the following parameters: (a) fever, (b) splenomegaly, (c) cytopenia (at least bilinear), (d) increased serum ferritin, (e) hypofibrinogenemia and/or hypertriglyceridemia, (f) increased soluble CD25 (soluble IL-2 receptor), (g) presence of haemophagocytes in the bone marrow, lymph nodes or spleen, (h) decreased or absent NK-cell activity.7,23 Some scores have been proposed by the scientific community and may be used for a successful risk assessment process to suspect HLH; for example, the HScore uses some of the parameters recommended by HLH-2004 and could help the clinician in the early detection of HLH. 24 In the primary or familial HLH form the diagnosis can be made by identifying mutations in at least one of the HLH-correlated genes or in presence of affected siblings.
Despite the proposed criteria, the diagnosis is still a challenge for both clinicians and pathologists, due to the low frequency of the disease and the complexity of interpreting both the clinical symptoms and laboratory findings. To complete the clinical picture, it is essential to verify the presence of hemophagocytes in the bone marrow biopsy or bone marrow blood aspirates, although haemophagocytes may also be occasionally present in other pathologies. In fact, the presence of haemophagocytosis without HLH-specific laboratory biomarkers may be associated with different pathological conditions. Among these disorders, haemophagocytosis has been reported to be present in infections, in onco-haematological conditions, in beta-thalassaemia and different types of anaemias.25,26 In particular, haemophagocytosis of mature red blood cells may be found in transfusion-dependent anaemia. 27 In addition, macrophage erythrophagocytosis has been reported to be pivotal in the resolution of inflammation, in particular in haemorrhagic inflammation.28,29 Even though haemophagocytosis is still recognized as usually associated with HLH, no conclusive correlation has been established between the number or type of cells phagocytized in the bone marrow and HLH. 30
There have been attempts to define quantitative and qualitative morphological parameters to categorize HLH haemophagocytes in the bone marrow of patients affected, but no shared outcomes have been achieved.30,31 Furthermore, in these studies the majority of patients were either adults or had a very wide age range 31 and included few or no primary cases. Paediatric cohorts might present some differences not only because of the age, but also for the presence of primary HLH forms. Therefore, in this work we retrospectively analysed blood parameters and haemophagocytes in a cohort of paediatric patients affected by HLH (including primary cases) and controls, with a focus on the lineage of cells ingested by haemophagocytes, in order to better define their possible utility in the diagnosis of HLH in children.
Material and methods
Subjects
This retrospective study considered all paediatric patients followed in the ASST Spedali Civili of Brescia Children’s hospital between 2001 and 2023 for whom a bone marrow smear analysis was requested to the Haematology section of the Clinical Chemistry Central Laboratory with the indication of ‘suspect HLH’. The following additional inclusion criteria were also applied to define a cohort of potential patients: (a) age at diagnosis less than 18 years, (b) presence of a complete medical record, (c) availability of laboratory tests at the onset of symptoms and before the start of any therapy.
Biographical, clinical and laboratory data were collected. Patients whose final diagnosis was not HLH were considered as controls.
Methods
Morphological bone marrow examination
Slides with bone marrow blood smears collected at the time of the disease onset and archived in the Haematology section of the Clinical Chemistry Laboratory were recovered and evaluated. Samples that had already been stained and samples that had only been dried (back-up slides) were found. For most patients it was possible to retrieve slides already stained at the time of sample collection. For patients whose stained slides were no longer available, staining of a back-up slide was performed by using the May-Grunwald-Giemsa method (eosin, methylene blue, methylene azure).
Samples stored for more than 7 years were of insufficient quality because they had lost the typical differential staining that distinguishes red blood cells and leukocytes, no longer showing the pink/red colours given by eosin, in favour of the blue tint given by methylene blue. These samples were therefore excluded from further evaluation.
A light microscope was utilized by two haematopathologists, respectively, with four and thirty years of experience for blind evaluation of bone marrow blood smear slides (Olympus BX53, Tokyo, Japan) and representative images were submitted to digital camera acquisition (SC180 with the cellSens Entry v1.18 software, Olympus, Tokyo, Japan). Initially, the samples were studied at lower magnification (10× and 20× or 40×) to assess the quality of the preparation and to identify areas of increased cellularity or areas with increased presence of haemophagocytes. They were then analysed at high magnification (60× or 100×) to obtain the cell count. 1,000 intact nucleated cells were counted for each sample. The greatest possible number of cells was counted in the case of a low cellularity and a proportional calculation per 1,000 cells was made. The numbers of total histiocytes per 1,000 cells and haemophagocytes per 1,000 cells were obtained. Haemophagocytes were then subdivided according to the lineage of the phagocytized cells (mature erythrocytes, nucleated erythrocytes, neutrophilic granulocytes, lymphocytes, platelets). If haemophagocytes contained several different nucleated cells at the same time, their percentage was also calculated out of 1,000 intact nucleated cells.
Laboratory testing
The biomarkers evaluated were among those included in the HLH-2004 criteria (haemoglobin, platelet count, absolute number of neutrophil granulocytes, fibrinogen, blood triglycerides and blood ferritin); in addition to these, the lactate dehydrogenase (LD) was also evaluated. CBC was performed by Sysmex XN-9000 instrumentation (Sysmex Corporation, Kobe, Japan); triglycerides, LD and ferritin were analysed by Roche Cobas 8000 (Sarstedt, Nümbrecht, Germany); fibrinogen was evaluated using Siemens CS-5100 instrumentation (Siemens Healthineers, Erlangen, Germany).
All available biomarkers between disease onset and before the start of therapy were collected. Reported values were the minimum (nadir) values of haemoglobin (g/L), platelets (109/L), neutrophil granulocytes (103/µL) and plasma fibrinogen (g/L); the maximum (zenith) values of plasma triglycerides (mmol/L), plasma ferritin (µg/L), plasma LD (U/L).
For patients and controls HScore was calculated as referred in [24].
Statistical analysis
Data were analysed using Stata Statistical Software (Stata Corp, Texas, USA) and GraphPad Prism v.5.0 (GraphPad Software, Massachusetts, USA). Proportions were compared by Fisher’s exact test. Quantitative variables in HLH cases and non-HLH controls were compared using the non-parametric Mann-Whitney test. ROC (Receiver Operating Characteristic) curve analysis was performed to determine the diagnostic accuracy of each biomarker in discriminating HLH cases from controls. The best cut-off values were calculated by the Youden index. A P-value <.05 was considered significant.
Results
36 potential patients were initially identified to be included in the study according to the clinical suspect of HLH. Among them, 20 patients fulfilled all the inclusion criteria. Only 1 patient was subsequently excluded because it was impossible to evaluate the bone marrow preparation due to inadequate staining, and therefore 19 patients were included in the study. These patients were divided into two groups according to the diagnosis reported in their medical record: 11 were HLH patients (cases) and 8 were patients with other haematological conditions but not affected by HLH (controls).
Patients and controls characteristics.
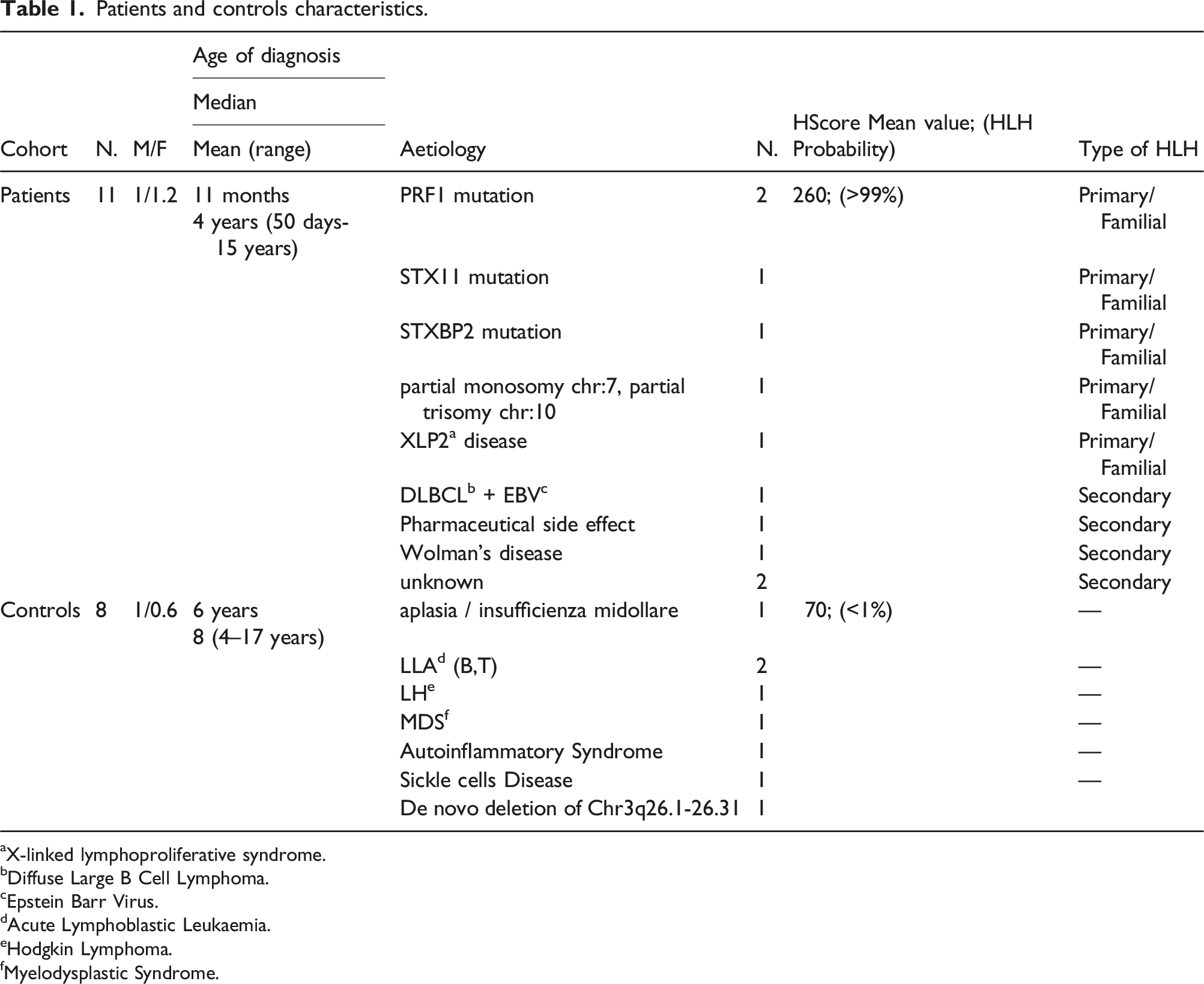
aX-linked lymphoproliferative syndrome.
bDiffuse Large B Cell Lymphoma.
cEpstein Barr Virus.
dAcute Lymphoblastic Leukaemia.
eHodgkin Lymphoma.
fMyelodysplastic Syndrome.
A familial or primitive HLH form affected about half of the patients (6 patients). Specifically, mutations in PRF1 (2 patients), STX11 (1 patient) and STXBP2 (1 patient), lymphoproliferative disease linked to chromosome X type 2 (XLP2) (1 patient), and polymalformative syndrome with an unbalanced chromosome structure with partial monosomy of chromosome 7 and partial trisomy of chromosome 10 (1 patient) were found. For secondary forms (5 patients) the aetiology was: unknown (2 patients), EBV-positive diffuse large B cell non-Hodgkin lymphoma (DLBCL) (1 patient), iatrogenic from anti-epileptic therapy with carbamazepine (1 patient), Wolman’s disease (1 patient). Patients in the control group were characterized by haematological diseases without HLH (8 patients) (Table 1). Almost all primary HLH-affected children were under 1 year of age. Therefore, HLH cases resulted significantly younger than controls. No statistically significant difference was found between the ages of primary HLH versus secondary HLH.
The blood biomarkers haemoglobin, platelet count, absolute number of neutrophil granulocytes, fibrinogen, blood triglycerides, blood ferritin and LD were evaluated in patients and in controls (Figure 1). Biochemical markers values. The figure showed the comparison among blood biochemical markers obtained in HLH patients and controls. P-values were obtained by using Mann-Whitney non-parametric test.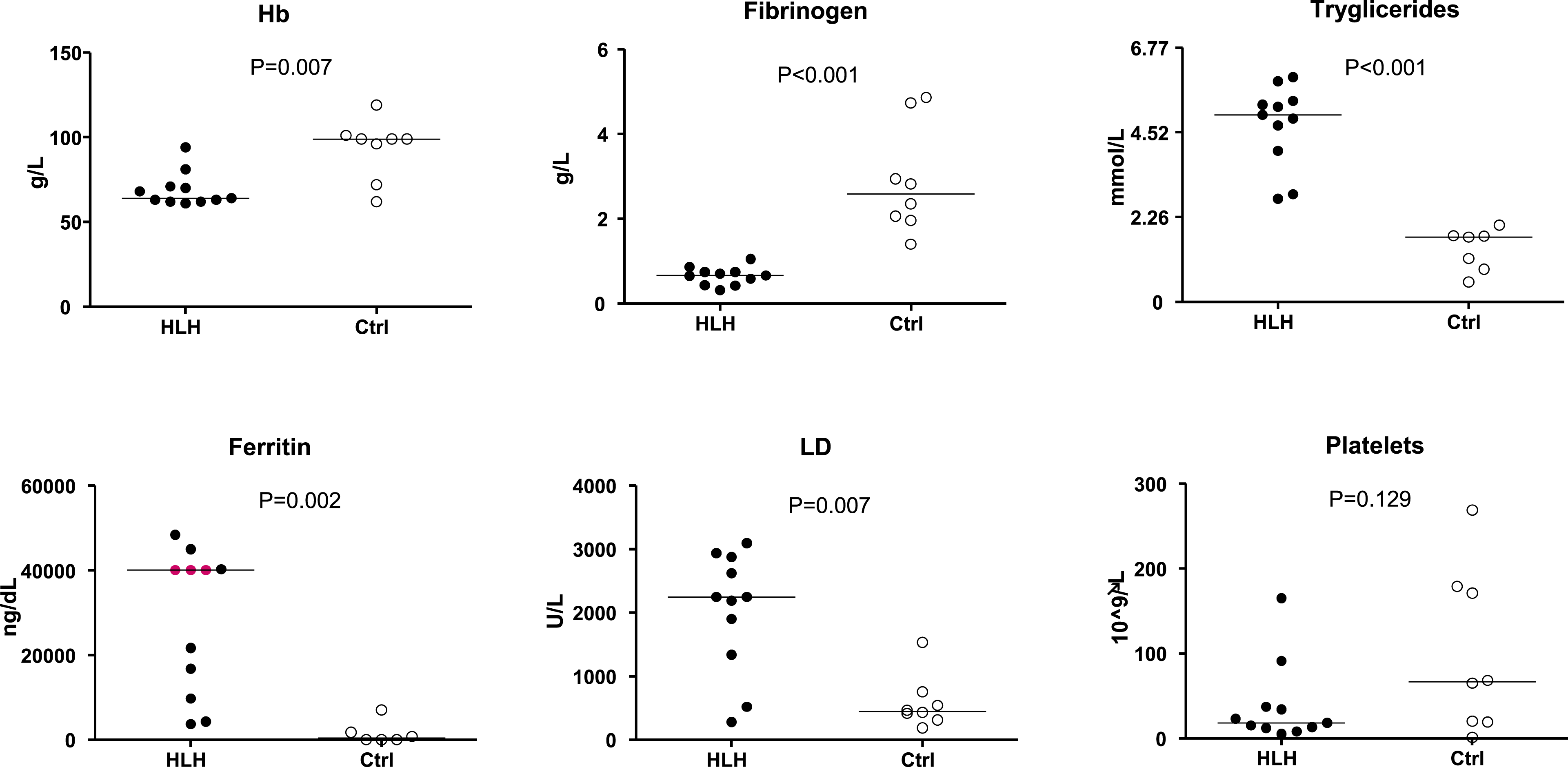
The comparison of the values resulted statistically significant for all the parameters except for platelet count (platelet count HLH patients vs controls: mean, median, range, respectively: 38 vs 99 × 109/L, 18 vs 66.5 ×109/L, range 5-165 vs 1-269 ×109/L). Haemoglobin and fibrinogen resulted lower in HLH than in controls (Haemoglobin HLH patients vs controls: mean, median, range, respectively: 69 vs 93 g/L, 64 vs 99 g/L, range 61–94 vs 62–119 g/L, P = .007; Fibrinogen HLH patients vs controls: mean, median, range, respectively: 0.649 vs 2.89 g/L, median 0.66 vs 2.585 g/L, range 0.31–1.05 vs 1.40–4.86 g/L, P < .001), while blood triglycerides, serum ferritin and LD were increased in HLH (Triglycerides HLH patients vs controls: mean, median, range, respectively: 4.71 vs 1.39 mmol/L, 4.98 vs 1.73 mmol/L, range 2.74–5.98 vs 0.53–2.04 mmol/L, P = .001; serum ferritin HLH patients vs controls: mean, median, range, respectively: 28,163 vs 1,625 µg/L, 40,000 vs 450 µg/L, range 3,721–48,368 vs 16–7,029 mg/dL, P = .002; LD HLH patients vs controls: mean, median, range, respectively: 2,023 vs 580 U/L, 2,248 vs 447 U/L, range 283–3,091 vs 187–1,531 U/L, P = .007) (Figure 1). The only significant differences we found between the primary HLH cases and secondary forms were a lower ferritin concentration (mean: 19095 vs 39043 µg/L; median: 13,267 vs 40,212 µg/L; range 3,721–40,000 µg/L vs 21,664–48,368 µg/L, P = .030) and a slightly lower haemoglobin (mean: 63 vs 76 g/L; median: 63 vs 71 g/L, range 61–68 vs 63–94 g/L, P = .022) in primary cases. Of note, 5 out of 6 secondary cases had a high serum ferritin concentration (≥ 21,664 mg/dL).
The performance of biochemical tests in discriminating patients and controls was evaluated by ROC curve analysis for all the biomarkers dosed. Blood triglycerides and fibrinogen discriminated HLH cases from controls perfectly, showing a sensitivity and specificity of 100% (AUC = 1) using the cut-off values of triglycerides ≥ 274 mmol/L and fibrinogen ≤ 1.40 g/L. Ferritin showed a sensitivity of 100% and a specificity of 83% (AUC 0.96) with the cut off ≥3,721 µg/L. LD was found to have a sensitivity of 73% and specificity of 100% (AUC 0.87) with the cut off ≥1,903 U/L. Haemoglobin was found to have a sensitivity of 75% and a specificity of 100% (AUC 0.87) with the cut off ≤ 96 g/L. (Figure 2). ROC curve of significant biochemical parameters. Significant P-values ROC curves with corresponding AUC Depicted in red: cut-off values chosen to maximize Youden’s index, which captures a test performance.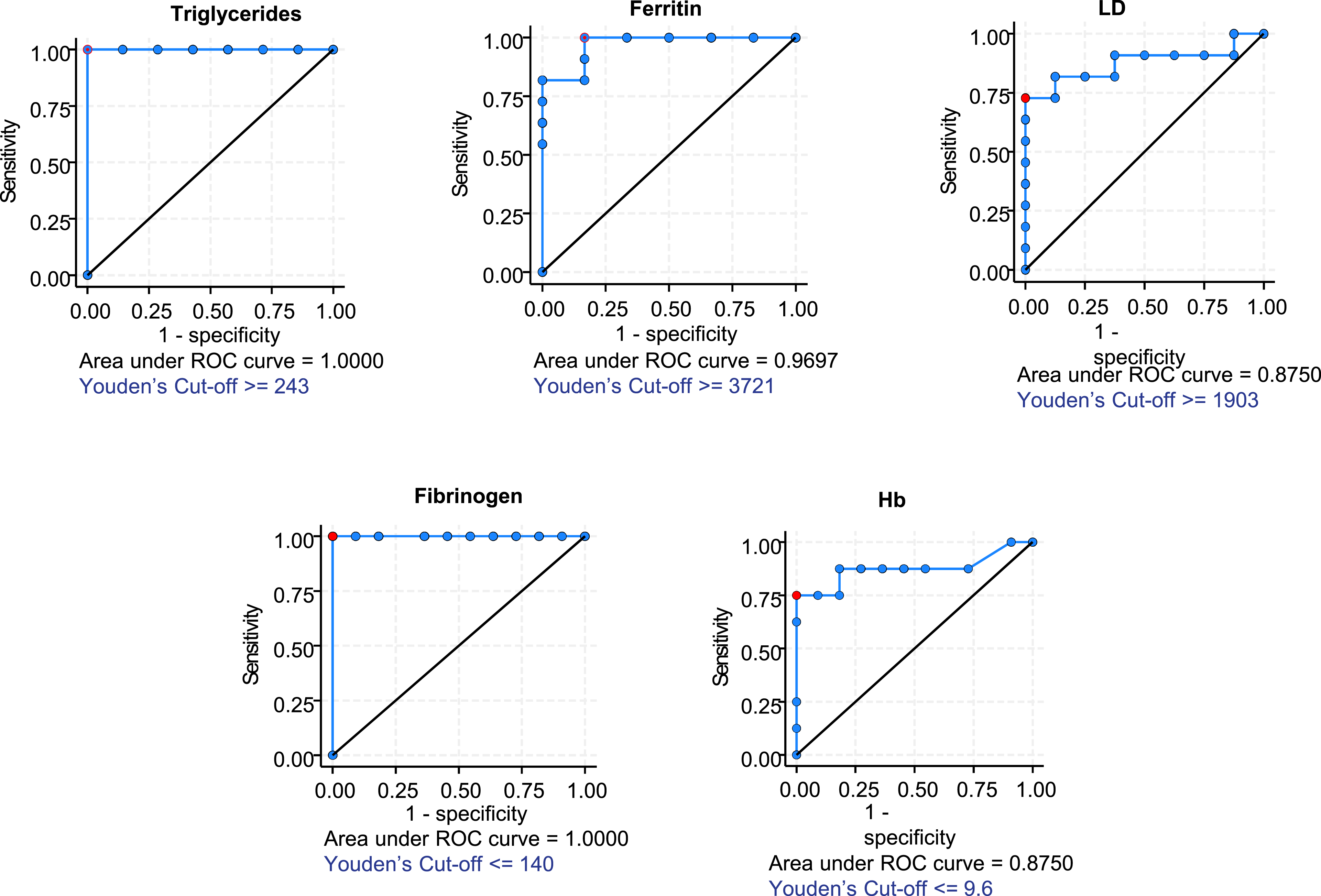
The cell count of 1,000 intact nucleated cells was obtained by analysing bone marrow blood smears. Histiocytes and haemophagocytes that engulfed mature erythrocytes, platelets, neutrophil granulocytes, lymphocytes, nucleated erythrocytes or total haemophagocytes were considered in this evaluation (some examples of the observed cells are reported in Figure 3. The cell counts of patients and controls were analysed and compared but, in most cases, they failed to demonstrate any statistically significant difference. Only the finding of increased phagocytosed NRBC was found to be statistically significant in the HLH cohort vs controls (mean: 1.8 vs 0 NRBC/1,000 cells, median: 1 vs 0 NRBC/1,000 cells, range 0–8 vs 0–0 NRBC/1,000 cells; P = .016), while a non-significant trend (P = .088) was found for increased phagocytosed erythrocytes (Figure 4(a)). In these two cases, the diagnostic accuracy of the cell counts was evaluated. Examples of microscopic views. (a) Bone marrow smear, (b) histiocyte containing hemosiderin and cellular debris; (c) haemophagocyte containing one erythroblast, some erythrocytes and some platelets; (d) haemophagocytes containing neutrophils, erythrocytes and platelets. Haemophagocytes. (a) Comparison between HLH patients and controls of NRBC and erythrocyte haemophagocytosis. (b) Significant P-values ROC curves with corresponding AUC Depicted in red: cut-off values chosen to maximize Youden’s index, which captures a test performance.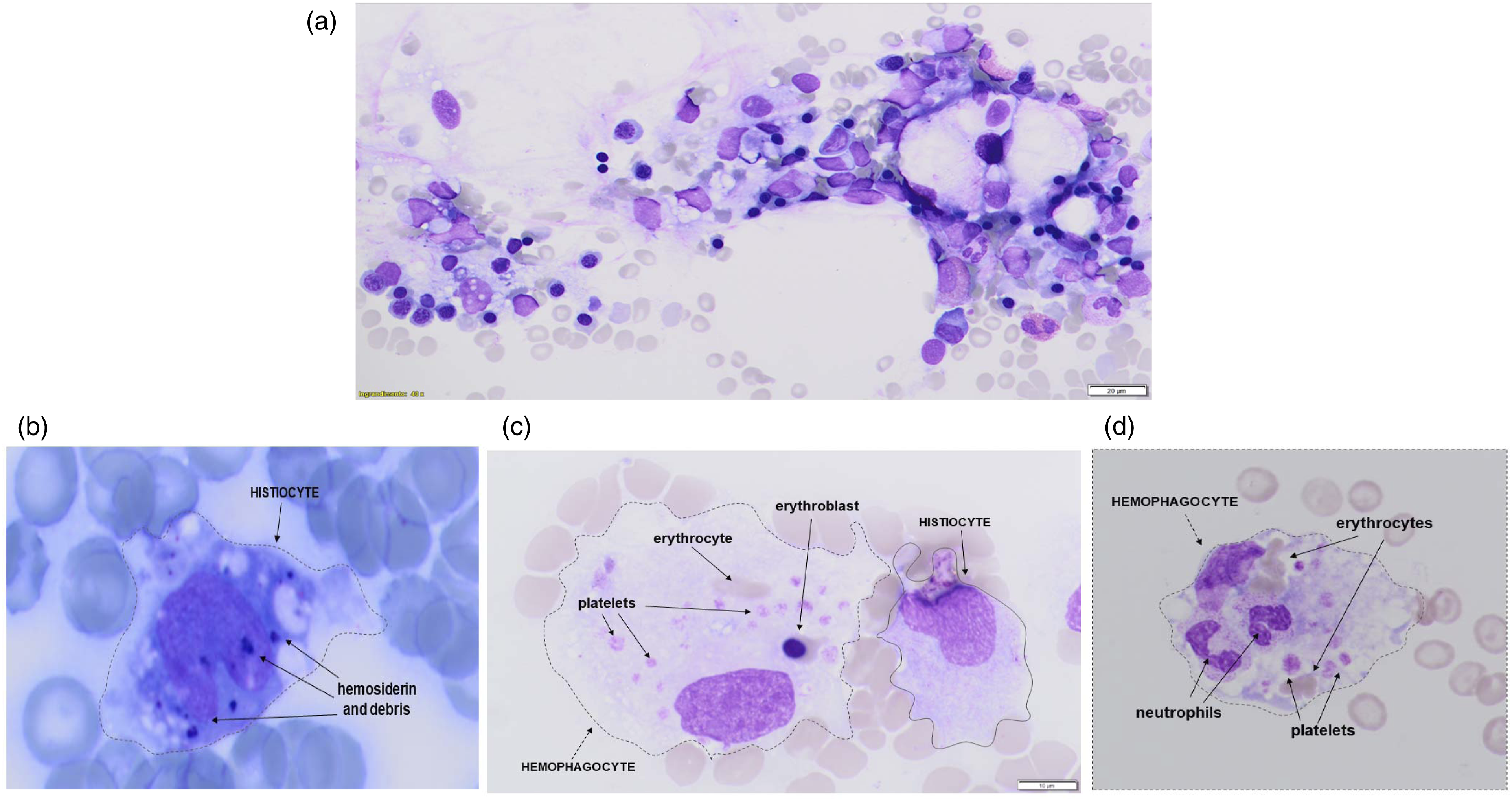
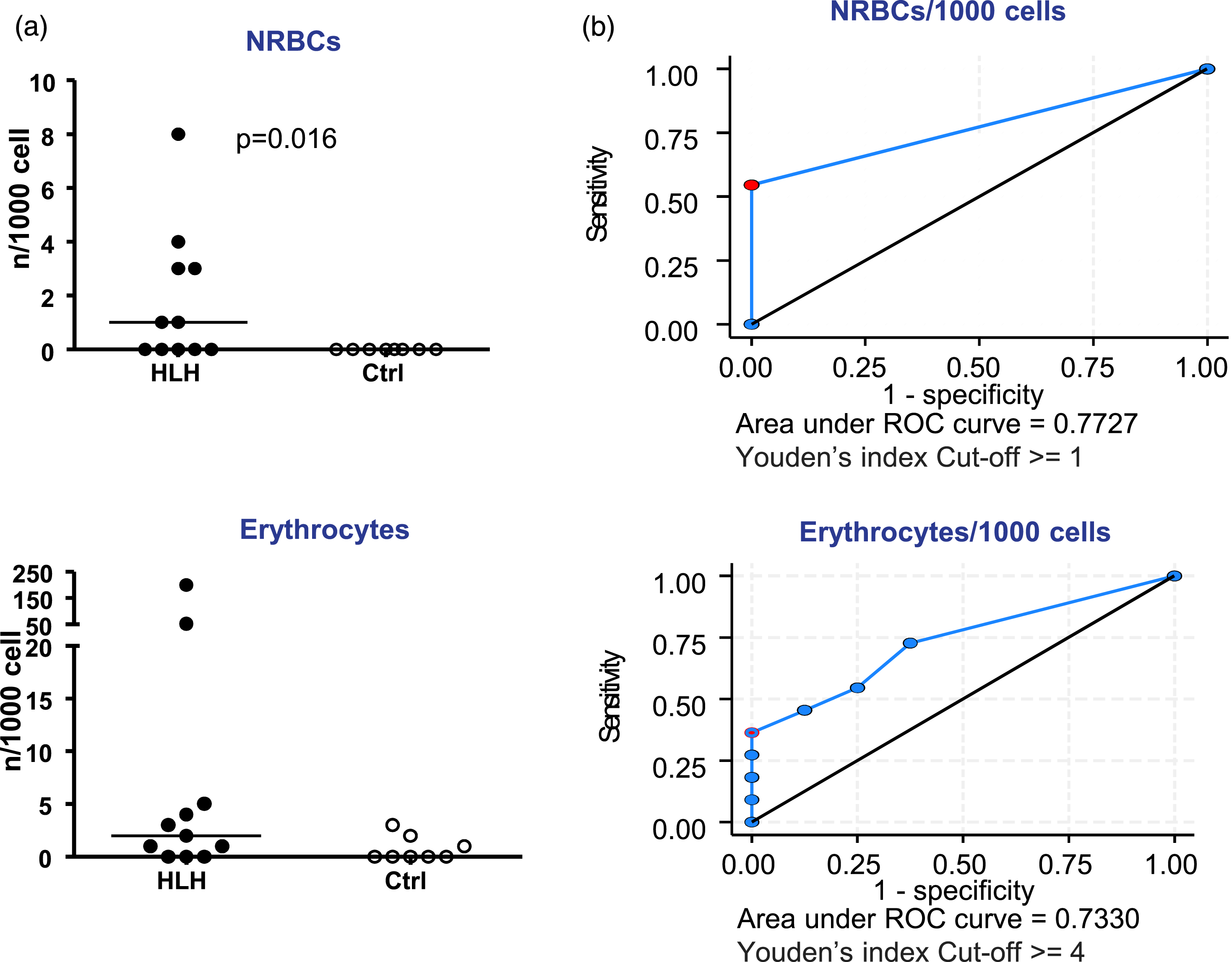
When NRBC/1,000 cells were considered, the sensitivity and specificity were, respectively, 55% and 100% with a cut-off of at least 1 phagocytosed cell (AUC = 0.77). In the case of erythrocytes/1,000, the sensitivity and the specificity were, respectively, of 36% and 100% using a cut-off value of at least 4 phagocytosed cells (AUC = 0.73) (Figure 4(B)). Of note, phagocytosed NRBC were more numerous in secondary HLH cases (NRBC median: 3 vs 0, range 0–8 vs 0–1, P = .043).
Finally, when considering haemophagocytosis of white blood cells, it was only observed in two secondary HLH cases and in two controls. One of the HLH cases showed 8 phagocytosed granulocytes/1,000 cells and the other one showed 1 phagocytosed lymphocyte/1,000 cells, whereas in controls there were 2 phagocytosed granulocytes/1,000 cells in one subject and 1 phagocytosed granulocyte/1,000 in the other.
Discussion
Since HLH is a severe disease with a high mortality rate especially when unrecognized, it is essential to identify HLH promptly to start the appropriate therapy. One of the criteria provided by the HLH-2004 guidelines is the presence of haemophagocytes in the bone marrow. However, the presence of these cells is often relatively non-specific and unlike other biochemical markers, explicit criteria for histo-morphological evaluation of haemophagocytes in HLH have not yet been rigorously established. Some researchers tried to classify the haemophagocyte types in relation to the lineage of cells phagocytosed with the aim of increasing the power of HLH diagnostic criteria, but their cohorts were mainly or entirely made up of adult patients and the results remained unsatisfactory.26,30–32
The specificity of haemophagocytosis was analysed by Ho et al., defining the absolute number of haemophagocytes in the bone marrow smears of adult patients affected by secondary HLH. In this work, the presence of haemophagocytes did not show specificity even when they were present in high numbers. 26
Bone marrow blood aspirates were examined by Gars et al. in 40 HLH patients in a very wide age range (from 10 months to 96 years) with only 1 primary HLH. The Authors observed that phagocytosis of non-nucleated erythrocytes alone was a non-specific finding, whereas haemophagocytes engulfing granulocytes, nucleated erythrocytes and at least one haemophagocyte containing multiple nucleated cells were strongly associated with HLH. The simultaneous haemophagocytosis of granulocytes, nucleated erythrocytes and lymphocytes allowed them to discriminate patients affected by HLH from non-affected ones. 31
Wilson et al. analysed the bone marrow specimens of 59 adult HLH patients. The Authors observed a significant association between the number of haemophagocytes and the other parameters provided by the HLH-2004 criteria. The haemophagocytes were present in about half of the patients studied and a significant correlation between the number of haemophagocytes and the values of serum ferritin was found. 32 24 patients were analysed in a recent work including 6 patients under the age of 18 years, with no primary HLH cases. The Authors showed that, among the HLH-2004 criteria, only hypertriglyceridemia had a significant association with HLH and that haemophagocytes with engulfed granulocytes were more represented in HLH cases. The Authors concluded that hypertriglyceridemia and the increased presence of haemophagocytes engulfing granulocytes may be predictive of HLH. 30
In our study, we assessed both biochemical and morphological parameters in a cohort composed of paediatric HLH patients, including both primary and secondary forms, in order to evaluate the diagnostic accuracy of the tests in the diagnostic process. In addition to the criteria included in HLH-2004, we also reported LD concentration and platelet haemophagocytes.
The best diagnostic performance was obtained with plasma triglycerides and fibrinogen, which allowed us to discriminate HLH cases from controls perfectly (100% sensitivity and specificity). The cut-offs were: triglycerides ≥ 2.74 mmol/L (very similar to HLH-2004 which indicates ≥ 2.99 mmol/L) and fibrinogen ≤ 1.40 g/L (≤ 1.50 g/L according to HLH-2004). Haemoglobin and LD also evidenced a good accuracy using the cut-off value of haemoglobin ≤ 96 g/L (≤ 90 g/L for HLH-2004) and LD ≥1903 U/L. Even though LD is not required by the HLH-2004 criteria, LD was often tested in our HLH patients, similarly to what reported in other cohorts of patients.33–35 It has already been reported that serum ferritin is an important marker in paediatric HLH with values >10,000 µg/L showing a 90% of sensitivity and 96% of specificity.36,37 In our small cohort, we found a lower ferritin cut-off value ≥3,721 µg/L (HLH-2004 indicates ≥500 µg/L) with a diagnostic performance of 100% sensitivity and 83% specificity. Ferritin was particularly high in secondary cases. In addition, we discovered that the HScore performed well in ruling in or ruling out HLH in our small cohorts, but it is rarely used by our Clinicians and the score still rely heavily on the HLH-2004 criteria, presence of haemophagocytes in the bone marrow included.
Among the morphological parameters of the bone marrow blood smear, the best diagnostic performance was achieved by the finding of haemophagocytes engulfing mature and nucleated red blood cells (RBC ≥4/1,000 cells; NRBC ≥1/1,000 cells). But in terms of sensitivity, their performance was less than that of blood biomarkers. Platelet haemophagocytosis did not demonstrate diagnostic significance. Two samples of patients affected by HLH resulted extremely cytopenic and almost devoid of nucleated cells other than macrophages. In one of these cases, however, haemophagocytes containing platelets were extremely represented (70/1,000 cells). Although the results of the present study do not show a statistically significant difference, we consider that further research on platelet haemophagocytosis in a larger population of patients may be interesting. We could therefore not replicate the results reported previously by Yan et al., 30 which showed a high specificity for granulocyte haemophagocytosis, because these haemophagocytes were found in only one of our secondary HLH children and in our cohort the presence of just 1 phagocytosed NRBC/1,000 was already highly specific for HLH in contrast to the 4 NRBC indicated by Gars et al. 31 . In addition, it is important to note that the finding of haemophagocytosis can help to diagnose HLH, but in the early phase of the disease this sign could be absent. 36 In particular, it is useful to remember that macrophage erythrophagocytosis is also present during the resolution of inflammation, especially in case of haemorrhagic inflammation.28,29
Our study has some limitations. First, because HLH is a rare disease and the cohort of HLH patients we could identify was numerically limited. This has determined a low statistical power that may have influenced the non-significance of some results obtained in the cell counts. The poor preservation of bone marrow aspirate samples over time and the low cellularity of the samples may also have contributed to reducing the quality of the morphological part of the study. In addition, the low power of the statistical tests could have had an impact on the comparability between primary and secondary forms, which prevented us from reaching a definitive conclusion about any characteristics that may be used to distinguish primary from secondary cases. However, the few differences we have found (lower ferritin and higher haemoglobin) further suggest that this topic should be worth exploring in larger studies. In addition, 3 HLH patients had serum ferritin concentrations that exceeded the guaranteed limit of linearity of the method (>40,000 µg/L) and the retroactive dilution of the samples to perform a precise measurement was not possible. For this reason, the mean values of serum ferritin in this cohort could have been underestimated. The blood biomarker soluble CD25 (soluble IL-2 receptor) is comprised in HLH-2004 criteria, and it is known to have a high sensitivity for HLH.38,39 However, it was not measured in our study because it is not a marker routinely tested in our laboratory and because our study was retrospective, and we had not stored the original blood samples. Finally, a blind morphological examination of the bone marrow blood smears was performed to limit the operator bias, so intra- and inter-operator variability influencing cell counts were not assessed; in this context, further studies may be necessary to verify the magnitude of the potential statistical error that might be introduced. Because of the very small cohorts of patients and controls, the results obtained should be taken as indicative. For this reason, an enlargement of the sample size by performing a multicentre study or the development of prospective studies could provide more robust data. Despite the study limitations, some of the cut-off values we obtained for the laboratory tests show a very strong diagnostic accuracy in our paediatric cohort and are very similar to those indicated by HLH-2004.
In conclusion, the achievement of shared criteria to classify haemophagocytes based on the lineage of the engulfed cells would be desirable. The results of our study urge the need for dedicated studies that are designed for paediatric populations and divided into primary/familial or secondary forms. Based on the results obtained in our small case study, we would recommend paying great attention to the morphological parameters and, among them, nothing could be neglected. We showed in our data that also haemophagocytosis of mature and nucleated erythrocytes could be significant. Therefore, the role of haematopathologists may be crucial to add precious information for HLH diagnosis because the construction of indices combining haemophagocytes morphology and blood biomarkers could be a promising and useful approach to HLH diagnosis.
Footnotes
Acknowledgements
The authors would like to thank God for enabling them to work for those people who suffer from health problems.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by Ex60% prof. Biasiotto of the University of Brescia.
Ethical approval
This study was performed according to the declaration of Helsinki. According to Italian law, since this study relies completely on pre-existing data acquired for diagnostic and therapeutic reasons, approval is no longer required.
Guarantor
Prof. Giorgio Biasiotto.
Contributorship
Elisa Caravaggi (EC), Federico Serana (FS), Mattia Carini (MC), Fabiana Ferrari (FF), Daniela Tregambe (DT), Moira Micheletti (MM), Giovanni Martellosio (GM), Duilio Brugnoni (DB), Roberto Bresciani (RB), Giorgio Biasiotto (GB). Conceptualization: EC, FS, RB, GB. Methodology: EC, FS, FF, MM, RB, GB. Investigation: EC, FS, DT, GM, GB. Visualization: FF, MM, RB, GB. Funding acquisition: GB. Project administration: GB. Supervision: FS, MM, GB. Writing–original draft: FS, MC, FF, GB. Writing–review & editing: EC, FS, MC, FF, DT, MM, GM, DB, RB, GB.