
Abstract
Background
Factors associated with interindividual variability in the pharmacokinetics of micafungin have been identified. This variability can cause underexposure and loss of drug efficacy. For this reason, a simple, fast, cost-effective and sensitive ultra-performance liquid chromatography ultraviolet detector (UPLC-UV) method was developed and validated for the quantification of micafungin.
Methods
The method involves simple plasma precipitation by UPLC with a reversed phase C18 column at 40°C coupled with ultraviolet detection set at a wavelength of 264 nm. The mobile phase consisted of a mixture 42/58 of potassium phosphate 20 mm and acetonitrile.
Results
The method was validated over the concentration range of 0.25–15.0 mg/L and proved to be reliable and reproducible with an average percentage of recoveries of 101.59 ± 3.93% and inter and intraday variation coefficients lower than 15% in all cases. The method was successfully applied in determining 30 samples from 10 patients being treated with micafungin.
Conclusions
The method proposed could be useful to facilitate the implementation of therapeutic drug monitoring for personalizing micafungin treatment in invasive fungal infections.
Keywords
Introduction
Invasive fungal infections (IFIs) have shown a significant increase during the last few decades. 1 Despite advances in the treatment of these infections, it is currently an important cause of morbidity and mortality worldwide, particularly in hospitalized and immunosuppressed patients,2,3 with a significant impact in costs for the public healthcare systems.
Micafungin is an antifungal drug from the echinocandins group with indication for the treatment of invasive and oesophageal candidiasis, in addition to the prophylaxis of Candida infections that are frequently observed in immunosuppressed patients. 4 It has fungicidal activity against most of Candida species, including azole resistant strains. In addition, it significantly inhibits the growth of filamentous fungi of Aspergillus species. 5 Although current guidelines recommend echinocandins as first-line therapies for most types of invasive candidiasis, 3 microbiological resistance to this antifungal agent has emerged and can result in clinical treatment failure.
Unlike other antifungals such as azoles, little evidence supports the routine use of therapeutic drug monitoring (TDM) for optimization of micafungin dosages. 6 However, some authors have identified several factors, such as age, body weight, SOFA score, platelet count and albumin level associated with interindividual variability in the pharmacokinetics (PK) of micafungin.7–12 This variability can cause underexposure to the drug in some patients such as neonates, pediatrics, obese subjects, patients in critical condition or those with haematological diseases when standard recommended dosage regimen is administered. Underexposure of the drug can cause loss of efficacy and development of fungal resistance,13,14 which could justify the need for TDM in certain groups of patients.
Several HPLC method with UV or fluorescence detection and methods using liquid chromatography-tandem mass spectrometry (LC-MS/MS) have been developed for the quantification of micafungin in plasma,15–19 which could help to obtain a deep understanding of the relationship between drug exposure and treatment response to micafungin, necessary to perform TDM.
The aim of this study was to develop and validate a simple, rapid and inexpensive ultra-performance liquid chromatography ultraviolet detector (UPLC-UV) method to measure plasma concentrations of micafungin and to investigate its clinical suitability for TDM in routine clinical practice.
Materials and methods
Chemicals
Micafungin pure drug substance was provided by Sigma-Aldrich® laboratory. HPLC grade acetonitrile was supplied by Thermo Fisher Scientific, KH2PO4 was purchased from Sigma-Aldrich® laboratory and ultrapure water was obtained with a Wasserlab Automatic Plus System.
Standard solution, calibration standards and quality control sample
Stock solution of 250 mg/L of micafungin was prepared in acetonitrile. Calibration standards and quality control (QC) samples in plasma were prepared diluting the corresponding stock solution with blank human plasma. Final concentrations of the calibration standards were 0.25, 0.5, 1.0, 5.0, 10.0 and 25 mg/L and the concentration of the QC were 0.25, 1.0, 5.0 and 25 mg/L. All stock solutions, calibration standards and QCs were immediately stored at −20°C.
Sample preparation
Blood samples were collected in EDTA tubes obtained from patients on micafungin therapy. Immediately after drawing, blood samples were centrifuged (3700 rpm for 3 min) and two plasma aliquots were collected into labelled polypropylene Eppendorf tubes stored at −18°C, up to a maximum of 3 months.
Frozen plasma samples were thawed and vortex-mixed before use. A total of 100 μl of acetonitrile was added to 100 μl of plasma samples and vortexed for 5 min and centrifuged for 5 min at 5000rpm. Subsequently, supernatant was filtered through 0.22 μm (Millex® syringe filters PVDF) and 10 μl were injected. The extraction efficiency of the direct plasma deproteinization method, verified in previous studies in the laboratory, was 96, 98 and 102% at 0.5, 10 and 25 μg/ml of micafungin, respectively, against the water.
Chromatographic conditions
A Waters Acquity High Pressure Chromatographer was used for quantifying micafungin with UV (UPLC-UV), performing chromatographic separation with a Luna Omega C18 column (1.6 μm; 2.1 mm × 50 mm, Phenomenex Company) with the following conditions: 0.5 mL/min flow, injection volume of 10 μL, 2 min of analysis time (1.3 min micafungin retention time) and 40°C column temperature. An UV scan of micafungin gave a λmax of 264 nm, thus this wavelength was used to analyse the drug. The mobile phase used for elution consisted of a mixture 42/58 of potassium phosphate 20 m
Method Validation
The method for quantification of micafungin in human plasma was validated for selectivity, linearity, intra and interday precision and accuracy, lower limit of quantification (LLOQ), lower limit of detection (LOD), carry-over and stability criteria according EMA Guideline for bioanalytical method validations. 20
Selectivity
Blank human plasma from five different healthy donors was processed and analysed to determine whether endogenous plasma peaks co-eluted with micafungin. During the analysis of the samples, a peak purity study was carried out.
Linearity
To evaluate the linearity, three calibration standards of eight concentrations of micafungin (0.25–15.0 mg/L) were analysed. The obtained data were used to build a calibration curve by taking the peak area versus the concentration of the sample. A 1/x2-weighted linear, least squares regression model was chosen to fit the calibration curve and slope and intercept were calculated with SPSS (IBM SPSS Statistics for Windows, Version 27.0. Armonk, NY)
LOD and LLOQ
The LOD was defined as the lowest concentration with a signal-to-noise ratio of ≥3. The lower limit of quantification (LLOQ) was defined as the lowest point in the calibration curve determined with acceptable precision and accuracy.
Intra and interday precision and accuracy
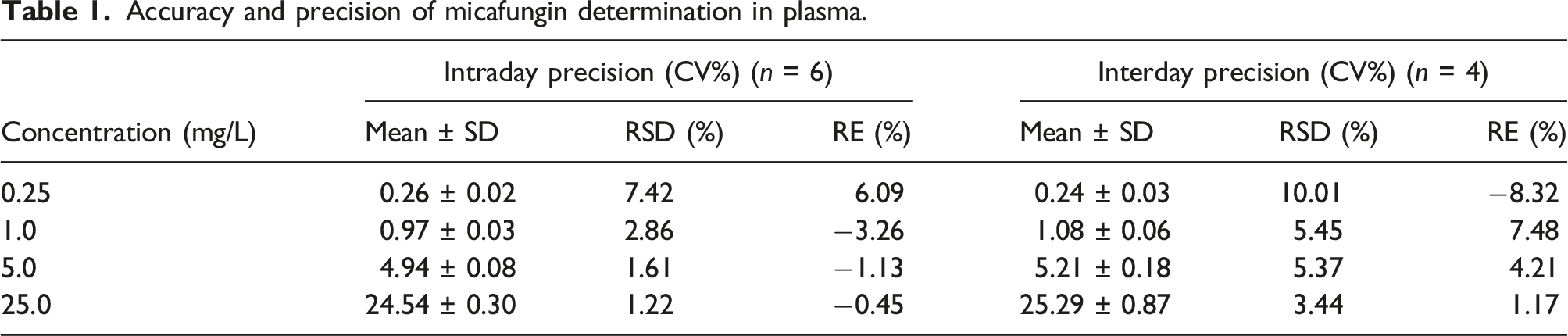
The precision and accuracy of the method were assessed by determination of four QC samples in plasma at different concentrations (0.25, 1.0, 5.0 and 25 mg/L) on four separate days. Precision was expressed as the percentage of relative standard deviation (RSD) and accuracy was expressed as the percentage of relative error (RE) between the measured and the nominal value. The precision for QC samples should be lower than 15% and the accuracy between −15% and 15% over the entire calibration range. A t-test for paired samples, assuming non-homogenous variances, was performed for the RE recovery values of the accuracy analysis.
Carry-over
Sample carry-over was measured using blank samples (n = 3) and standard calibrator at the upper limit of quantification (25 mg/L) for micafungin plasma concentrations. After the injection of a high plasma concentration, a blank plasma sample was injected, and the carry-over was calculated as the percentage of mean peak area at LLOQ for micafungin. Carry-over for the analyte was accepted if the signal was less than 20% of the signal of the LLOQ.
Sample stability
Drug stability in plasma was assessed using low (0.25 mg/L) and high (25 mg/L) calibration standards under different conditions, such as at room temperature for 8 h, after three freeze-thaw cycles, stored at −20°C for 3 months and at room temperature at 30, 60, 90 and 120 min after sample preparation. Samples were considered stable if assay values were within the acceptable limits of accuracy (RE<±15%) and precision (RSD<±15%).
Clinical suitability
To explore the technical applicability of the method in the clinical setting, micafungin concentrations were measured in plasma samples obtained from patients who received micafungin as prophylaxis for IFIs. Micafungin was administered as an intravenous infusion over a period of 30 min at a dose of 50 mg per day.
The extraction of the samples was carried out on the fifth day after the allogeneic haematopoietic stem cell transplant. The sampling times were 0.5 h, 3 h and 24 h after finishing the infusion. The blood samples were collected in EDTA-flushed tubes, centrifuged at 3700 rpm for 3 min and stored at −20°C until analysis. The steady-state 24-h area under the concentration curve (AUC0-24) was calculated by the linear trapezoidal rule using the total micafungin concentration-time data.
Taking into account that the most accepted value for the Pharmacokinetic-Pharmacodynamic (PK/PD) parameter for the most prevalent Candida spp. in our environment is an AUC/MIC >3000 mg·h/L, 8 and assuming a mean of minimal inhibitory concentrations (MIC) of 0.016 mg/L5, the cut-off point for AUC0-24 to maximize the efficacy of the antifungal treatment was set at 50 mg·h/L. Moreover, a cut-off point for the minimum concentration (Cmin) target of micafungin was established at 1 mg/L derived from in vitro susceptibility tests of Candida spp. 6
Ethics
The study protocol was authorized by the Ethics Committee of Clinical Research of the local University Hospital of Salamanca (CEIC number: PI2021/02/685), and all patients signed informed consent regarding their willingness to participate in the study.
Results
Method validation
Linearity
The peak area ratio versus the nominal concentration of micafungin showed a linear relationship over the concentration range of 0.25–25.0 mg/L in human plasma. A linear regression equation for the calibration curve resulted in the following equation: Y = 101.6 + 1767.9x and a correlation coefficient (r) of 0.9999. The LLOQ in human plasma was established in 0.25 mg/L with RSD and RE of 7.42% and 6.09%, respectively.
Selectivity
Figure 1 shows representative chromatograms of blank drug-free plasma sample, a plasma sample spiked with micafungin (5.0 mg/L) and a patient sample. No endogenous plasma peaks were observed at the retention time of the studied analyte. No interferences with co-administered drugs (e.g. levofloxacin, omeprazole, ondansetron, tacrolimus, sirolimus or mycophenolate mofetil) were observed. Lack of interfering peaks with the micafungin in any sample was shown in the peak purity study (purity angle < purity threshold). Representative chromatograms of (A) drug-free plasma sample, (B) drug-free plasma spiked with 5.0 mg/L of micafungine and (C) patient sample.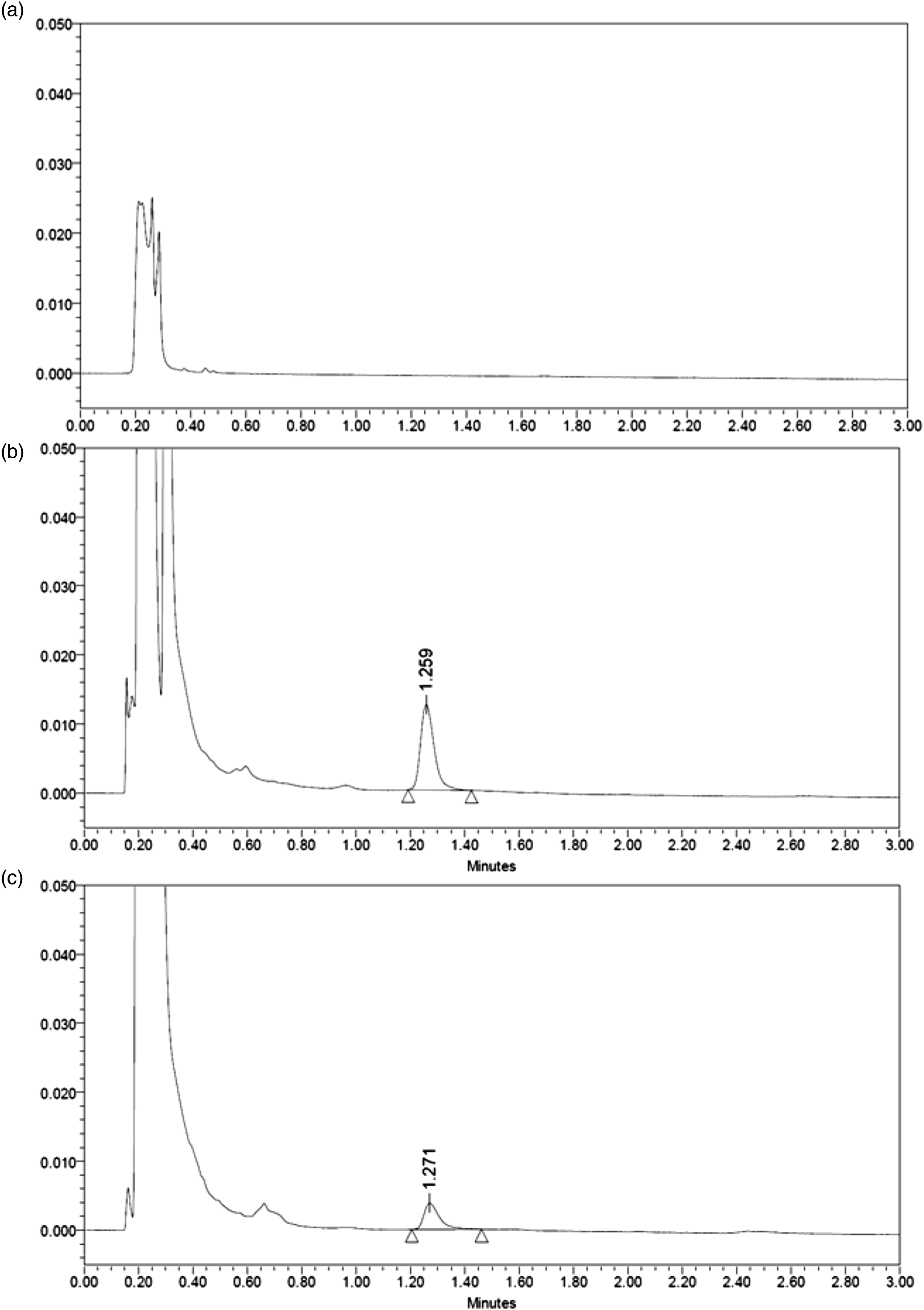
Precision and accuracy
Accuracy and precision of micafungin determination in plasma.
Carry-over
An absence of significant carry-over phenomenon was demonstrated as the mean carry-over (±SD) expressed relative to the response at the LLOQ, was 9.57% ± 4.52%, value below the acceptance criterion proposed by the EMA guideline (<20% at the LLOQ).
Sample stability
Stability results of micafungin in human plasma in different conditions (n = 3).

Clinical suitability
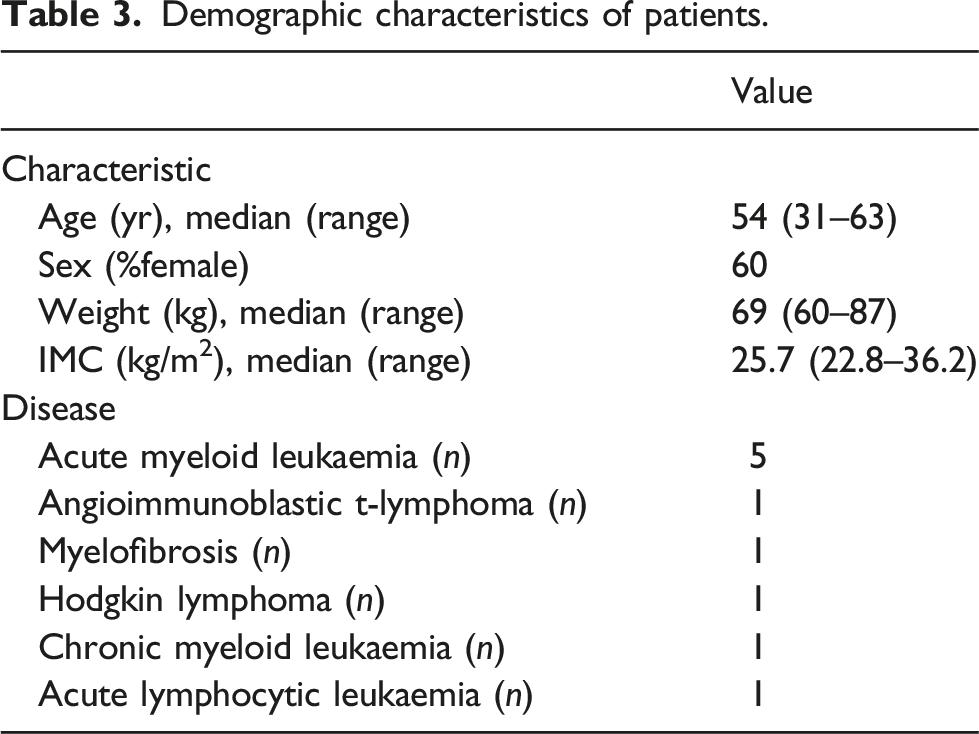
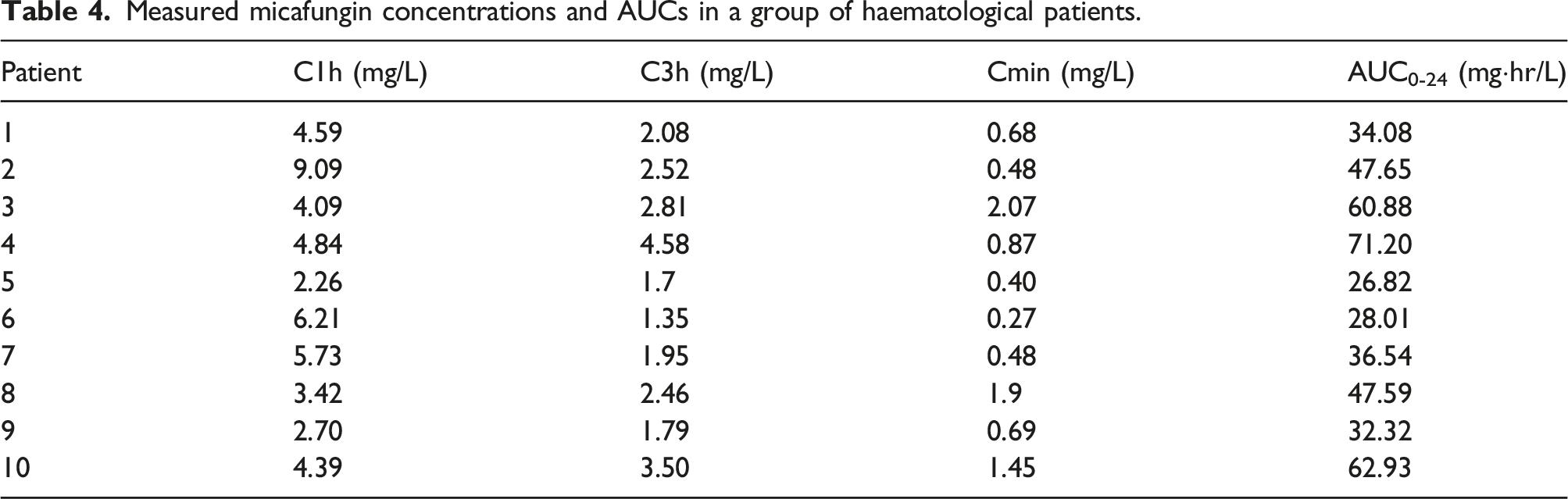
The clinical suitability of the method was evaluated by analysing 30 plasma samples obtained from 10 patients undergoing allogeneic haematopoietic stem cell transplant who received prophylaxis for IFIs with micafungin from day 1 post-transplant. Micafungin was detected in all samples and the levels quantified were within the concentration ranges established in the new UPLC-UV method presented in this work (0.27–9.09 mg/L).
Demographic characteristics of patients.
Measured micafungin concentrations and AUCs in a group of haematological patients.
Discussion
Micafungin is an antifungal drug with concentration-dependent fungicidal activity against most of Candida species. It has been previously reported that equinocandin under exposure compromises the efficacy of treatment and may be the cause of resistance development. 6 Although little evidence supports the routine use of TDM for optimization of micafungin dosage, exposure monitoring should be considered for patients in whom greater pharmacokinetic variability has been observed. The following target patient populations for TDM have been proposed: neonates, children, adolescents, obese subjects, those with hepatic or renal impairment, critically ill individuals and patients with haematological diseases. 6
In view of the substantial disease burden associated with IFIs, more robust evidence is required in order to determine the clinical utility of micafungin TDM for dosage optimization to increase the likelihood of therapeutic success and to minimize any potential toxicity. One of the main challenges for applying this tool involves availability of a reliable and accessible analytical method to quantify micafungin in plasma.
Chromatographic techniques coupled to mass spectrometry detection have been previously proposed for the quantification of micafungin in plasma, all of them characterized by high sensitivity and specificity.15,18,19 However, these methods are time consuming and require highly trained staff and expensive equipment which limits implementation in clinical laboratories. In contrast, one of the main advantages of our UPLC-UV method over other currently available chromatographic techniques is the fast sample preparation and chromatographic run, which allows for processing up to 20 samples per hour. Moreover, taking into account only direct costs, the estimated price of each determination is approximately $3US per sample. These characteristics make this method accessible and affordable for implementation in clinical practice.
The analytical method presented in this work was successfully validated for the selectivity, linearity, precision, accuracy and stability criteria according to EMA recommendations on bioanalytical method validation in the expected range of micafungin concentrations in the treatment of IFI. The LLOQ of the developed UPLC-UV method, 0.25 mg/L, is similar to most of already published chromatographic methods. 17 It is true that some methods coupled to an MS detector make it possible to work with lower LLOQs, 19 although taking into account that the lower limit of the proposed micafungin trough concentration therapeutic range is 1 mg/L, 6 the LLOQ established for the developed UPLC-UV is sufficient for daily clinical practice for micafungin TDM.
Bioanalytical validation should demonstrate that the method is reliable and reproducible for its intended use. We applied our method to determine micafungin in 30 samples of 10 patients being treated with this drug. All samples were correctly quantified, with concentrations above the LLOQ within the validation range. In our study, all patients received the standard regimen dosage of micafungin according to the drug data sheet recommendations, with high variability observed in the serum concentrations of the drug. This may indicate a significant difference in the pharmacokinetic behaviour of this drug in the haematological population, which could be at risk of underexposure and treatment failure. These preliminary results support the usefulness of the developed analytical method for TDM purposes to personalize treatment with micafungin.
The method developed presents the limitations that only micafungin is measured and no potential interfering micafungin metabolites are quantified. The simultaneous detection of two active metabolites of micafungin is widely described in the literature.12,21 The metabolites, M1 (catechol form) and M2 (methoxyform), are known to be therapeutically potent treatments for experimental infections. However, Kenji Tabata et al. 12 considered that due to their low plasma concentrations, these metabolites have no therapeutic relevance.
Conclusion
We developed and validated a fast and sensitive UPLC-UV method for quantification of micafungin. Simple sample preparation and short acquisition time enable a high sample throughput while being cost-effective. The method was successfully used for analysis of patient samples demonstrating its applicability in a clinical setting. It provides the basis for further investigations on therapeutic drug levels and concentration-dependent effects and may be used for pharmacokinetic and pharmacodynamic studies of micafungin.
Footnotes
Acknowledgements
We would like to thank the patients, and the medical, nursing and laboratory staff who participated in the clinical studies included in the present work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
The study protocol was authorized by the Ethics Committee of Clinical Research of the local University Hospital of Salamanca (CEIC number: PI2021/02/685), and all patients signed informed consent regarding their willingness to participate in the study.
Guarantor
None.
Contributorship
N.R. and A.Z.C. designed the study. A.Z.C. performed the micafungin quantification in plasma. M.M and N.R. performed the pharmacokinetic analysis, interpreted the data and wrote the manuscript. A.Z.C., N.R., J.S.P.B. and M.J.O. critically reviewed the manuscript. All authors approved the final version of the manuscript.