
Abstract
Autosomal recessive hypercholesterolemia (ARH; OMIM #603813) is an extremely rare disorder of lipid metabolism caused by loss-of-function variants in the LDL receptor adapter protein 1 (LDLRAP1) gene, which is characterized by severe hypercholesterolaemia and an increased risk of premature atherosclerotic cardiovascular disease. We report the case of an 11-year-old girl who presented with multiple painless yellowish papules around her elbows and knees of two-year duration. She had been reviewed by several general practitioners, with some of the papules having been excised, but without a specific diagnosis being made. The child was referred to a paediatric service for further evaluation and treatment of the cutaneous lesions, which appeared xanthomatous in nature. A lipid profile showed severe hypercholesterolaemia. Next generation sequencing analysis of a monogenic hypercholesterolaemia gene panel revealed homozygosity for a pathogenic frameshift mutation, c.71dupG, p.Gly25Argfs*9 in LDLRAP1. Her parents and brother, who were asymptomatic, were screened and found to be heterozygous carriers of the LDLRAP1 variant. There was no known consanguinity in the family. She was commenced on the HMG-CoA reductase inhibitor, atorvastatin, to good effect, with a ∼76% reduction in LDL-cholesterol at a dose of 50 mg per day. At six-month follow-up, there had been no obvious regression of the xanthomata, but importantly, no enlargement of, or the development of new papular lesions, have occurred. In summary, we report a child who presented with multiple cutaneous xanthomata and was confirmed to have ARH by the presence of a homozygous novel pathogenic frameshift variant in LDLRAP1.
Introduction
Autosomal recessive hypercholesterolemia (ARH; OMIM #603813) is an extremely rare inherited form of hypercholesterolaemia, with a prevalence of less than 1 per million people, caused by loss-of-function variants in the LDL receptor adapter protein 1 (LDLRAP1) gene. ARH is characterized by severe hypercholesterolaemia, due to a markedly elevated LDL-cholesterol (>10 mmol/L), and an increased risk of premature atherosclerotic cardiovascular disease (ASCVD). 1 LDLRAP1 codes for a cytosolic adaptor protein required for the hepatic LDL-receptor-mediated internalization and clearance of LDL particles. 2 Similar to autosomal dominant homozygous familial hypercholesterolaemia (hoFH), individuals with ARH may also present with cutaneous xanthomata, tendon xanthomata, progressive aortic stenosis and premature ASCVD.3,4
We report a child who presented with multiple cutaneous xanthomata, who was found to have severe hypercholesterolaemia and confirmed to have ARH by the presence of a homozygous novel pathogenic frameshift variant in LDLRAP1.
Clinical case
An 11-year-old previously healthy girl presented to a general practitioner with multiple painless yellowish papules located around both elbows and knees, which had initially appeared over her right elbow at age nine years and which had subsequently enlarged in size, increased number, becoming bilateral in distribution. Over the past two years, she had been reviewed by several general practitioners, with some of the papules having been excised for cosmetic purposes, but without a specific diagnosis being made. Her parents were not related and her father was known to be hypercholesterolaemic. The child was referred to a paediatric service for further evaluation of the cutaneous lesions, which were considered to be xanthomatous in nature (Figure 1). All other organ systems were normal by clinical assessment.
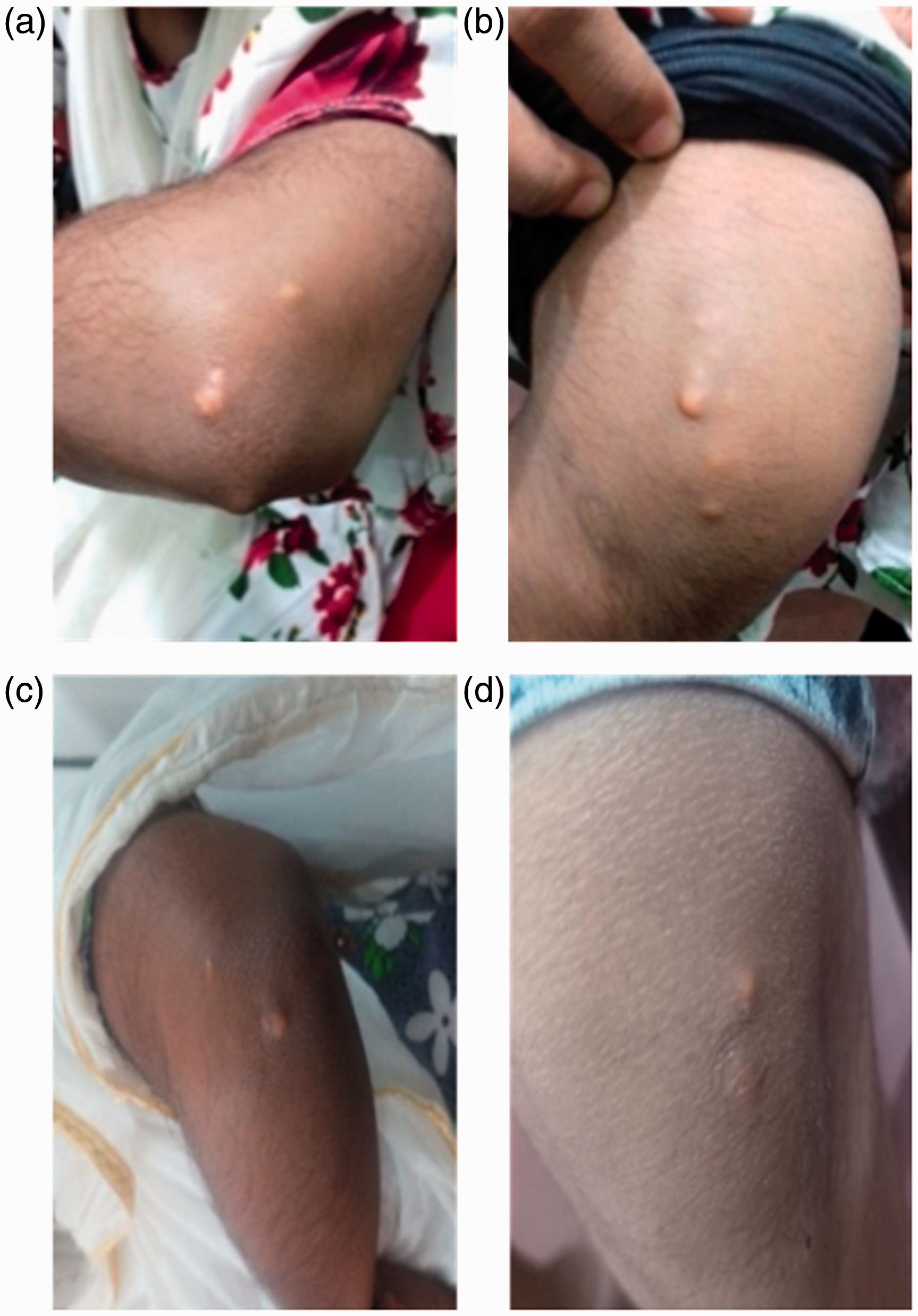
Bilateral cutaneous xanthomata over the elbows (a,b) and knees (c,d) of the ARH proband.
A lipid profile revealed severe hypercholesterolaemia, with total cholesterol and LDL-cholesterol concentrations of 17.3 mmol/L and 15.6 mmol/L, respectively (Table 1). Her full blood count, liver, renal and thyroid function tests were all within the reference intervals. We obtained DNA samples from the proband and three first-degree relatives (both parents and a brother) for molecular analysis. Informed consent was obtained from all subjects. DNA was extracted using a standard Triton X-100 salting out procedure. Next generation sequencing of a monogenic hypercholesterolaemia gene panel (LDLR, APOB, PCSK9, LDLRAP1, APOE, ABCG5, ABCG8, LIPA and CYP27A1) revealed homozygosity for a pathogenic frameshift LDLRAP1 variant, NM_01627.2:c.71dupG, p.Gly25Argfs*9. As expected, her parents were found to be heterozygous for the LDLRAP1 variant, as was her brother (Table 1). An electrocardiogram and 2D echocardiogram were both normal. She was commenced on the HMG-CoA reductase inhibitor, atorvastatin 30 mg daily, to good effect, with a ∼60% reduction in LDL-cholesterol (down to 6.0 mmol/L). Her parents were counselled about the natural history of ARH, the importance of compliance with statin therapy and the likely need for additional lipid-lowering treatment.
Lipid results of the ARH proband and her family.
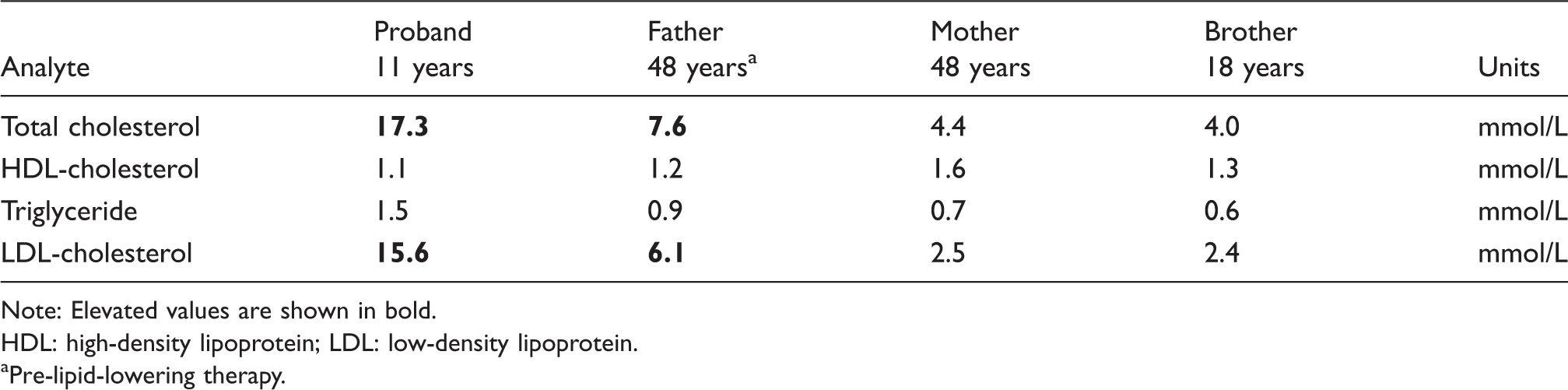
Note: Elevated values are shown in bold.
HDL: high-density lipoprotein; LDL: low-density lipoprotein.
aPre-lipid-lowering therapy.
Further biochemical testing was undertaken on the patient’s father to exclude secondary causes of hypercholesterolaemia, which showed normal renal function, liver function, thyroid function and glucose concentration. A cardiovascular examination was normal, as was an exercise stress test and 2D-echocardiography; there were no peripheral stigmata of lipid disorders. In order to exclude the possibility of heterozygous FH, the father’s DNA was also sequenced using the monogenic hypercholesterolaemia gene panel. No other pathogenic, or suspicious, variants were detected apart from the heterozygous LDLRAP1 frameshift variant. He is currently being treated with atorvastatin 10 mg once daily together with diet modification and other lifestyle measures for the management of his hypercholesterolaemia. Her mother and brother, who have a normal lipid profiles, report no cardiovascular symptoms and have no evidence of cutaneous xanthomata.
At follow-up of the patient, after six-months of statin therapy, there has been no regression of the xanthomata, but importantly, no enlargement of, or the development of new papular lesions, have occurred. She is currently receiving atorvastatin 50 mg per day, together with adherence to paediatric dietary guidelines as recommended by a dietician and now has an LDL-cholesterol of 3.7 mmol/L.
Discussion
ARH and hoFH are both characterized by severe hypercholesterolaemia (LDL-cholesterol >10 mmol/L), cutaneous xanthomata, tendon xanthomata, progressive aortic stenosis and premature ASCVD. 4 , 5 However, the clinical phenotype of ARH is more variable, and being generally regarded as less severe and more responsive to lipid-lowering therapy. Family screening is useful in differentiating between these conditions since obligate heterozygote parents of ARH patients typically have normal LDL-cholesterol concentrations, while parents of hoFH patients have heterozygous FH, with increased LDL-cholesterol concentrations (typically 5–10 mmol/L). Cutaneous xanthomata, due to cholesterol accumulation in the dermis, which can involve the dorsum of the elbows, knees, buttocks, ears, eyelids, hands and feet, may, as in our case, be the first indication of a life-threatening genetic lipid disorder.
ARH is caused by loss-of-function variants in the LDLRAP1 gene on chromosome 1p36.11. 2 LDLRAP1 encodes a 308 amino acid adaptor protein, LDLRAP1, which facilitates the endocytosis of LDL-receptors in clathrin-coated pits, particularly in polarized cells such as hepatocytes and lymphoblasts. The ARH protein includes three critical domains for LDL-receptor endocytosis: a phosphotyrosine-binding domain that binds to the C-terminal cytoplasmic tail of the LDL-receptor; a clathrin box domain which is required for the LDL-receptor to be recruited to clathrin-coated pits and an adaptor protein AP-2-binding region. 4 , 6
We have described a homozygous novel LDLRAP1 frameshift variant, c.71dupG (p.Gly25Argfs*9), in a child with severe hypercholesterolaemia, confirming a diagnosis of ARH. This variant is effectively a null allele, as translation of the protein is stopped before any functional domains can be produced. The majority of the ∼20 ARH-causing variants reported in the literature, including the first two reported variants from Sardinia (p.Ala145Serfs*26 and Trp22*), are nonsense, splicing, or frameshift mutations. 5
Our case demonstrates the classical features of ARH with the clinical findings of cutaneous xanthomata and severe hypercholesterolaemia, due to a markedly elevated LDL-cholesterol. 5 Although more common on the Italian island of Sardinia, 7 with an estimated carrier frequency of ∼1 per 143 of the population, 8 it has also since been described in a number of other countries, including Spain, Turkey and Japan, to name a few, 4 and relevant to our case, it has been previously reported in individuals of Asian Indian origin. 9 Although heterogeneous in nature, it has been observed, like in our case, that patients with ARH respond better to conventional lipid-lowering therapies, including high-dose statin and ezetimibe, than hoFH, with per cent reductions similar to that observed in the general population. 3 Consideration will be given to adding ezetimibe to our patient’s current lipid-lowering regimen to further improve her LDL-cholesterol concentration. However, it would seem likely that she will require additional measures such as PCSK9 inhibition and/or lipoprotein apheresis to achieve appropriate LDL-cholesterol targets.
As stated above, ARH can lead to multisystem complications and, if untreated, can have deleterious long-term effects; therefore, early and appropriate intervention is paramount. However, challenges exist with respect to diagnosis, best practice management and follow-up of the rare genetic lipid disorders, including ARH. 10 National and international patient registries will assist with increasing the awareness of and provide insight into ARH (and other rare genetic lipid disorders), the development of novel therapies and the integration of new knowledge to optimize clinical best practice and ultimately improve patient outcomes. 11 , 12
Conclusion
We report a child who presented with multiple cutaneous xanthomata, who was found to have severe hypercholesterolaemia and confirmed to have ARH by the presence of a homozygous novel pathogenic frameshift variant in LDLRAP1. The early diagnosis of ARH is paramount to prevent complications such as aortic stenosis, premature ASCVD and death. There is a need for patient registries to provide insight into the prevalence and natural history of ARH, improve diagnostic and management practices and enable clinical trials of new therapies.
Footnotes
Acknowledgements
We thank Ms. Lan Nguyen for her assistance with sequencing. This case report was presented in poster format at the 5th Annual Academic Sessions of College of Chemical Pathologists in Sri Lanka in 2020.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Written informed consent from the patient’s legal guardian was obtained.
Guarantor
VT.
Contributorship
VT and KD wrote the first draft of the article. All authors edited the article and approved the final version of the article.