
Abstract
Background and aim
Familial hypercholesterolaemia is caused by variants in the low-density lipoprotein cholesterol metabolic pathway involving LDLR, APOB and PCSK9 genes. A national genetic testing service in Wales, UK has observed that no familial hypercholesterolaemia variant is found in almost 80% patients with the familial hypercholesterolaemia phenotype. It has recently been suggested that some adult patients with a familial hypercholesterolaemia phenotype may have cholesteryl ester storage disease which can also present as a mixed hyperlipidaemia. The commonest genetic cause of cholesteryl ester storage disease is an exon 8 splice junction variant in the LIPA gene (rs116928232, c.894G>A; E8SJM) previously found to have an allele frequency of 0.0011 (1 in 450 individuals) in a large European population. This study investigated the prevalence of the E8SJM in patients with a familial hypercholesterolaemia phenotype in Wales, UK.
Method
A total of 1203 patients with a clinical suspicion of familial hypercholesterolaemia but no familial hypercholesterolaemia variant were invited to participate. Of these, 668 patients provided informed written consent. Stored DNA samples from 663 patients were genotyped for the E8SJM variant.
Results
Three heterozygotes were identified (allele frequency 0.0023). Whole gene sequencing of the LIPA gene was undertaken in these three individuals, but no other variants were found. Therefore, there were no cholesteryl ester storage disease patients (homozygote or compound heterozygote) identified in this cohort.
Conclusion
The allele frequency 0.0023 (1 in 221 individuals) for the E8SJM variant was more prevalent in this cohort than in a European population study; however, no cholesteryl ester storage disease homozygotes were identified. We found no evidence to support routine testing for cholesteryl ester storage disease in adult patients with a familial hypercholesterolaemia phenotype.
Keywords
Introduction
Familial hypercholesterolemia (FH) is an inherited condition characterized by lifelong exposure to elevated low-density lipoprotein cholesterol (LDL-C) and increased risk of premature coronary heart disease. Classical FH is caused by variants in the metabolic pathway for low-density lipoprotein (LDL), most commonly the LDL receptor gene (LDLR). FH can also be caused by variants in other genes involved in the metabolism of cholesterol, e.g. the APOB and PCSK9 genes. FH caused by variants in LDLR, APOB and PCSK9 behaves as an autosomal dominant monogenic disorder and is estimated to affect 1 in 250 of the general population in UK (approximately 262,000 people), although it is significantly underdiagnosed in clinical practice.1,2 In the UK and elsewhere, guidelines recommend genetic cascade testing from a genetically diagnosed proband to improve detection rates in younger patients so that they may be offered lipid-lowering treatment. 3 A very rare autosomal recessive form of FH caused by variants in the LDLRAP1 gene is also recognized. 4
The Wales FH Service
Wales has a well-established service for diagnosis and family testing of FH (since October 2010). This offers genetic testing to individuals with hypercholesterolaemia who present to lipid clinics across Wales on the basis of clinical signs, symptoms and family history. 5
Patients with a clinical diagnosis of possible or definite FH are offered genetic testing for common FH mutations. If a variant is found in an index patient, then family cascade testing is offered to relatives. Since October 2010, over 2000 index patients have undergone DNA testing for possible FH. A recognized pathogenic variant was found in approximately 21% of patients, and a further 7% of patients had genetic variants of uncertain significance (uncertain whether variant is pathogenic or non-pathogenic). 6 Seventy-two per cent did not have variants in any of the genes being tested, which is a similar value to studies of lipid clinic patients reported in the UK where the majority of patients tested had possible FH as defined by the Simon Broome criteria. 7 Individuals who do not have a pathogenic variant are managed with lipid-lowering treatment, but currently no further genetic testing is offered to their relatives. Their hyperlipidaemia is considered to be due to FH-causing variants in other genes not included in the test panel, for example in ABCG58,9or possibly in APOE 10 or in STAP1. 11 In the majority of cases, it is believed that the cause is polygenic in nature. 12
Cholesteryl ester storage disease
Cholesteryl ester storage disease (CESD) is a lysosomal storage disease that is also known as lysosomal acid lipase deficiency (LAL-D). In CESD, a deficiency of this enzyme leads to hyperlipidaemia and, in many patients, accumulation of lipids in the liver. There is a wide spectrum of severity, ranging from a severe early onset infantile form (Wolman Disease) through to a less severe, later onset adult form which is generally known as CESD. The majority of LAL-D patients present in childhood, 13 but there are some case reports of individuals presenting in adulthood with raised LDL-C similar to FH. 14
In CESD, the major problem is accumulation of cholesterol esters and triglycerides in lysosomes with multisystem effects including hepatic steatosis, hyperlipidaemia and premature atherosclerosis leading to early onset heart disease. 15 The frequency of shared symptoms with other conditions means that CESD is underdiagnosed. It has been reported that some patients with CESD may present with a lipid profile similar to that of FH.16,17 Standard lipid-lowering treatments have been shown to reduce the hyperlipidaemia in patients with CESD, but hepatic damage may still persist. 18 Therefore, it is important to accurately diagnose and treat CESD. Enzyme replacement therapy has been shown to effectively improve dyslipidaemia and liver abnormalities associated with LAL-D and has now been approved for use in both adults and children. 19
CESD is an autosomal recessive condition resulting from variants in the LIPA gene. The most common variant associated with CESD is an exon 8 splice junction mutation (E8SJM; c.894G>A), which encodes mostly inactive LAL with approximately 3–8% of normal activity. 20 This mutation has been estimated to account for 51%–69% reported variants among multiethnic CESD patients. 21
This variant was reported to have an allele frequency of 0.25% (1 in 200 persons) in a population cohort of 2023 German individuals enriched with patients attending Italian and German outpatient lipid clinics. 22 CESD heterozygotes in this study tended to have higher LDL cholesterol compared with controls and this reached statistical significance in men. A much larger study by Stitziel et al. reported the allele frequency to be lower: 0.11% (1 in 450 persons out of 27,472 tested) in individuals of European ancestry. 14 In contrast to the findings of Muntoni et al., they found no significant difference in lipids (LDL-C, Triglycerides [Tg], HDL-C) between individuals heterozygous for E8SJM and control subjects. 18 A recent Portuguese study found four LAL-D homozygotes within a sample of patients with a clinical, but not a genetic diagnosis of FH. All four patients were found to have the common E8SJM variant. 23
The current study aimed to determine whether the CESD (E8SJM variant) has been overlooked as a cause of hypercholesterolaemia in a Welsh cohort of adult patients with a clinical diagnosis of FH, but in whom the common FH genetic variants were absent.
Patients and methods
Participants were adult probands (≥18 years) with a clinical phenotype suggestive of familial hypercholesterolaemia who had undergone genetic testing for FH by the All Wales FH Testing Service and had a negative genetic test for FH. All participants provided written, informed consent for their stored DNA samples to be re-analysed for the current study. The study protocol conforms to the ethical guideline of the 1975 Declaration of Helsinki. A favourable ethical opinion was obtained from National Institute for Social Care and Health Research, Research Ethics Service: reference 14/WA/1008.
Stored samples were retrieved and aliquots sent to Cardiovascular Genetics, BHF Labs, Institute Cardiovascular Science, and University College London for analysis. DNA quantification was performed using the Nanodrop 800 (Thermo-Fisher Scientific, Loughborough, UK). The DNA was standardized in distilled water (Sigma-Aldrich (Gillingham, UK) to 15 ng/μL then a further dilution down to 2.25 ng/μL in Nunc polypropylene 96-well plates (Sigma-Aldrich, Gillingham, UK). A positive DNA control heterozygote for rs116928232; c.894C>T was used. Genotyping was performed using TaqMan technology (Life Technology, Thermo-Fisher Scientific, Loughborough, UK). TaqMan primer and probes were provided by Synageva BioPharma Corp, Lexington, MA, USA.
Further investigation of E8SJM heterozygotes
The coding region of the LIPA gene of identified c.984C>T heterozygotes was sequenced to check for the presence of another pathogenic variant to account for their phenotype. Primers were designed for sequencing the coding region and intron exon boundaries of the LIPA gene using Primer3 (http//www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). PCR was carried out using ACQUA PRC Mix (Applied Biosystems, Thermo-Fisher Scientific, Loughborough, UK) in a 20 μL final volume using a final concentration 0.4 pmol/μL of each primer on a Bio Rad C1000 thermal cycler. PCR products were verified by electrophoresis on a 1.5% agarose gel and subsequently purified by GFX column (illustra GFX PCR DNA and Gel Band Purification Kit, GE Healthcare, Cambridge, UK) before sequencing. Purified samples were sent to Source BioScience (Cambridge, UK) for sequencing. Subsequently, any amino acid changes observed were assessed in silico using Polyphen2, http://genetics.bwh. harvard.edu/pph2/, SIFT http://sift.jcvi.org/ and Mutation Taster (http://www.genomatix.de/matinspector.html)
LAL enzyme activity in c.984C>T heterozygotes was also determined from dried blood spots by the Biochemistry Department, Yorkhill Hospital, Yorkhill, Glasgow, UK. 24
Results
A total of 1203 FH variant-negative index patients were invited to participate. Of these, 668 provided written consent and 663 had a sufficient stored sample for analysis (Figure 1).
Of the 663 genotyped, only three were found to be c.984C>T heterozygotes.
Sequencing of the LIPA gene of these three participants showed that all three had one or more variants in exon 2: rs1051338 and rs1051339. However, these two variants are frequent and in the signal peptide (amino acids 1–23) which is cleaved in the mature protein. In silico assessment using Polyphen2, SIFT and Mutation Taster as shown (Table 1) indicates that these are not disease causing variants. Therefore, we conclude that none of the participants were found to have other variants in the LIPA gene that would give rise to compound heterozygosity.
Results of in silico assessment.

aPolyphen: (http://genetics.bwh.harvard.edu/pph2/index.shtml); SIFT: (http://sift.jcvi.org/www/SIFT_enst_submit.html); Mutation Taster: (http://www.mutationtaster.org/).
Dried blood spot analysis showed that all three heterozygotes had reduced LAL activity, typical of heterozygote carriers, but not in the significantly deficient range. All three patients had normal liver function tests. Lipid profiles (pre and on treatment) are shown (Table 2). The allele frequency of the E8SJM c.984C>T in this cohort was 0.0023 or 1 in 221.
Characteristics of c.984C>T heterozygotes.
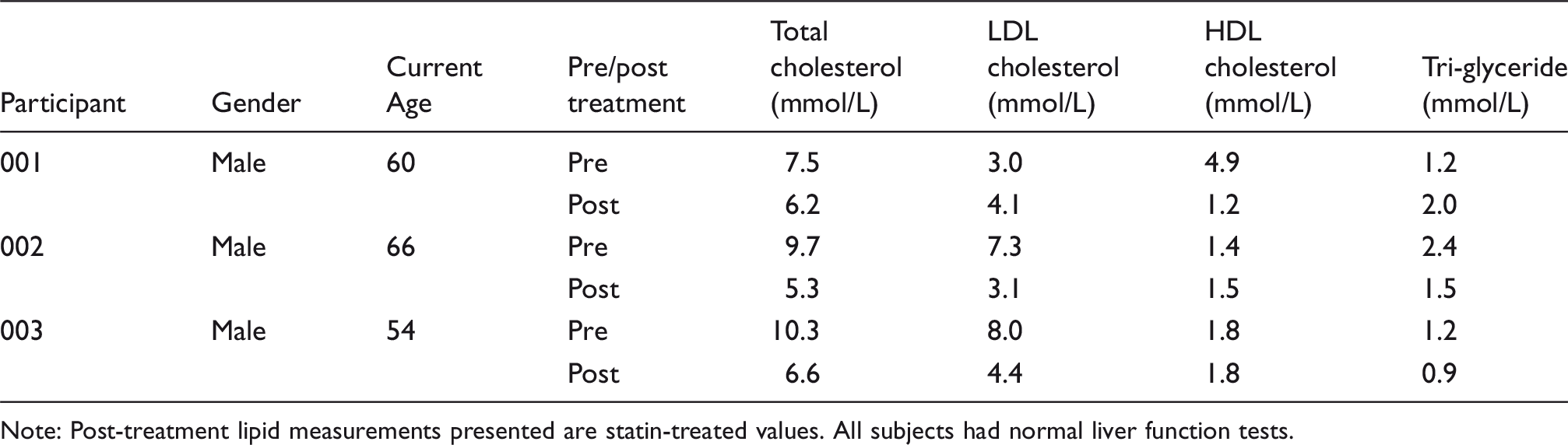
Note: Post-treatment lipid measurements presented are statin-treated values. All subjects had normal liver function tests.
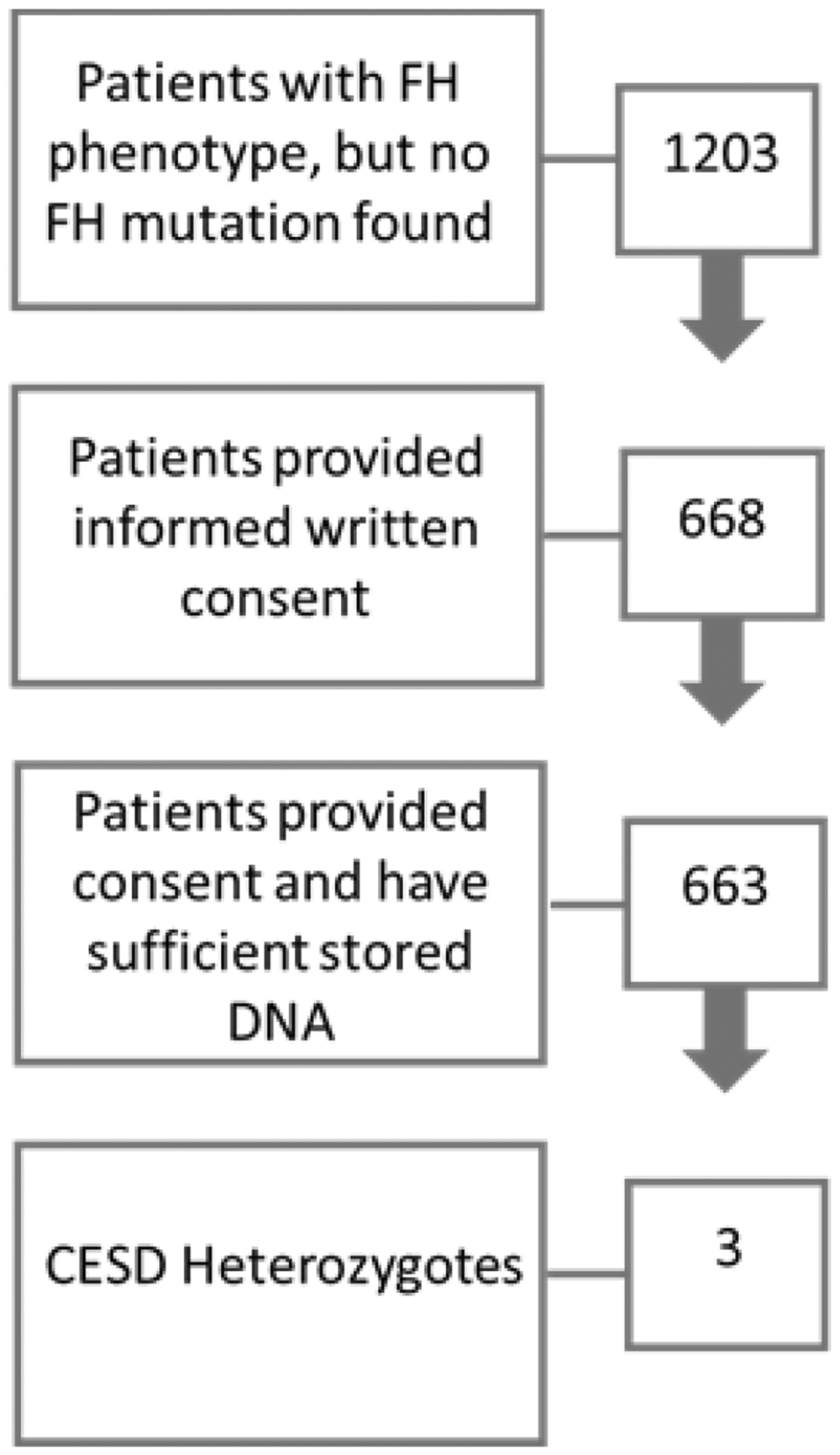
Numbers of patients at each stage of the project.
Discussion
This study was undertaken to investigate the hypothesis that CESD may be overlooked as a possible cause of lipid alterations in a cohort of patients with clinically suspected FH, but where no FH variant has been identified. There are examples of other genes that have been identified as causing an FH-like clinical presentation including ABCG5, APOE and STAP1 variants.8,10,11 The range of symptoms associated with CESD is sufficiently diverse that an overlap with FH and potential masquerading as this condition is conceivable and was first suggested by a European study which included a general population sample and hyperlipidaemic patients. 17 Later that same year, the suggestion of lipid alterations in E8SJM heterozygotes was challenged in a much larger European population cohort which reported no association of the E8SJM variant with either plasma lipid concentrations or MI/CAD risk and reported a lower carrier frequency of 0.16%. 14
In the current study, we have observed an allele frequency of 0.0023 or a carrier frequency of 1 in 221. This is similar to the observed allele frequency of 0.0025 in Muntoni’s German, population-based study. 22 Pullinger et al. used a similar methodology to the current study to determine the prevalence of the common E8SJM variant in 1357 patients with hyperlipidaemia in the USA. 25 These authors identified six CESD E8SJM heterozygotes (1 in 226), one of whom was found to be a compound heterozygote (frameshift variant involving an exon 4 deletion). A recent report from the Netherlands which included 213 adults and 63 children with a clinical, but no genetic diagnosis of FH, reported six heterozygous carriers of a potentially pathogenic variant in the LIPA gene. 26 Two of these patients were heterozygous carriers of c.894G>A variant. These authors reported finding no homozygous or compound heterozygous carriers of potentially pathogenic LIPA variants. A recent Norwegian study recently concluded that CESD was a very rare cause of hypercholesterolaemia when they observed only two patients with the E8SJM variant in a cohort of 3027 adult patients with an FH phenotype and none of the common variants in LDLR, PCSK9 and APOB. 27
In contrast to the studies discussed above, a recent Portuguese study of patients with a clinical diagnosis of FH (n = 492) and patients with dyslipidaemia and altered liver function (n = 258) identified 26 different variants in the LIPA gene. 23 These included 10 common polymorphisms identified in several individuals, 16 rare variants, 15 not described before in LAL-D patients, and three homozygotes with the c.894 E8SJM. All three E8SJM homozygotes were referred to the service as children and were in the FH variant-negative group. Following cascade screening, one child was found to have a homozygous sibling. No adults with the homozygous E8SJM were found. In all families with LALD patients, some, but not all carriers had altered lipid metabolism. This reflects the inconsistencies regarding the association between hyperlipidaemia and heterozygous E8SJM reported in previous studies. Moreover, this study illustrates the importance of establishing care pathways for unexplained hyperlipidaemia in children. In the absence of a genetic diagnosis of FH, it may be pertinent to consider dried blood spot testing for LALD as a diagnostic test especially if there are disturbed liver function tests and raised triglyceride.
The strategy of screening for the most common CESD variant (E8SJM) and subsequent full sequencing in carriers has been shown to be an efficient means of detecting carriers and combined heterozygosity at the LIPA locus. 25 Therefore, our results suggest that the CESD E8SJM does not appear to be enriched in this Welsh cohort of hyperlipidaemic patients with a clinical but no genetic diagnosis of FH. We conclude that testing for this variant in the LIPA gene is not clinically helpful in our cohort of adult FH patients and should not be added to our routine panel of genetic tests for hypercholesterolaemic patients with a phenotype of FH. It should be reserved for children with raised triglyceride and cholesterol with unexplained liver steatosis.
Highlights
An alternative diagnosis for the FH phenotype was investigated. A total of 663 no-mutation FH patients were examined. The common LIPA splice site mutation (E8SJM) was genotyped. Three carriers were identified for an allele frequency of 0.0023. CESD is not common in a Welsh cohort of patients with an FH phenotype.
Footnotes
Acknowledgements
We would like to thank the All Wales Medical Genetics Service for their help with processing the stored DNA samples.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Pauline Ashfield-Watt and Ian McDowell received funding from Synageva BioPharma in the form of a project grant as described below. Steve Humphries is the Medical Director and minority shareholder of a UCL spin-out company called StoreGene, which uses a 20 SNP genetic test, in combination with the classical risk factor profile, for estimating an individual’s future risk of CVD, and which offers FH genetic testing via an accredited DNA diagnostic laboratory. No other authors have declared any conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Synageva BioPharma (now part of Alexion Pharmaceuticals). This study was a researcher-led project. All elements of the design and execution of the study were led and undertaken by the researchers. The funder has not been involved in the preparation of this manuscript. SEH is supported by the British Heart Foundation (RG008/08) and by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Ethical approval
A favourable ethical opinion was obtained from National Institute for Social Care and Health Research, Research Ethics Service, Wales, UK: reference 14/WA/1008.
Guarantor
IMcD.
Contributorship
IMcD, PAW and KH researched the literature and developed the protocol. PAW and IMcD obtained ethical approval. PAW contacted the patients and co-ordinated sample retrieval and transport between labs. RE, DT and RG liaised with patients for additional study-specific blood samples. SEH and KWL analysed the samples. PAW drafted the manuscript and all authors reviewed it and approved the final version.