
Abstract
Thyrotoxic periodic paralysis is a rare complication of hyperthyroidism where increased influx of potassium into skeletal muscle cells leads to profound hypokalaemia and paralysis. Most cases arise sporadically in Asians; however, it is being increasingly reported in Caucasians. It is regarded as a channelopathy where a genetic and/or acquired defect in the sodium-potassium (Na/K-ATPase) pump renders it more sensitive to excess thyroid hormone in susceptible individuals. Because the clinical presentation is similar to familial hypokalaemic periodic paralysis, genes implicated in this autosomal-dominant condition became candidates for thyrotoxic periodic paralysis, particularly if they were known to have thyroid hormone-responsive elements. These include the voltage-gated calcium (CACNA1S) and sodium (SCN4A) channel genes, KCNJ18 which encodes the inwardly rectifying potassium channel Kir2.6, and subunits of the Na/K-ATPase genes. Although no single pathogenetic mutation has been identified in thyrotoxic periodic paralysis, several single-nucleotide polymorphisms in these genes have been associated with it. We describe a 27-year-old Caucasian Irish male who presented with acute onset limb paralysis and severe hypokalaemia. He was diagnosed as having thyrotoxic periodic paralysis secondary to Graves’ disease based on clinical presentation, biochemical findings and rapid response to intravenous potassium. Genetic analysis identified heterozygous variants in three candidate genes: KCNJ18 (c.576G>C), SCN4A (c.2341G>A) and CACNA1S (c.1817G>A). Since these variants are not disease causing and occur at high prevalences of 50%, 2–3% and 1%, respectively, in the normal population, they do not explain the clinical phenotype in our patient suggesting that acquired environmental triggers or as-yet unidentified gene mutations remain as leading pathogenetic co-factors in thyrotoxic periodic paralysis.
Case description
A 27-year-old Caucasian Irish male patient was brought to the emergency department with acute onset of generalized muscle weakness. He reported waking up that morning with severe leg weakness and was unable to walk. The weakness worsened rapidly to involve his upper limbs in ascending fashion which rendered him unable to move. He also complained of neck stiffness but denied sensory loss, fits, headache, blurring of vision, photophobia, abdominal pain, diarrhoea, urinary or respiratory symptoms. He also reported having a prior, but less intense episode of lower limb weakness six months previously that resolved spontaneously within an hour. He experienced intermittent palpitations and had lost about 10 kg in the previous six months despite increased appetite. He was not on any prescribed or over-the-counter medication and his medical history was insignificant. There was no history of similar illness in the family.
On physical examination, he was alert, oriented and generally unwell. His palms were sweaty. He was afebrile, blood pressure was 140/66 mm Hg, pulse 118 bpm regular, respiratory rate 30 breaths/min and oxygen saturation of 96% on room air. Neurological examination revealed proximal muscle weakness with power of 1/5, decreased muscle tone, diminished deep tendon reflexes in all four limbs and equivocal plantar reflexes bilaterally. His higher mental functions, sensory system and cranial nerves were intact. Cerebellar functions could not be assessed due to paralysis. There was no goitre and the rest of the systemic examination was within normal limits.
Laboratory studies (Table 1) revealed low serum potassium (K) of 1.7 mmol/L (reference interval, RI: 3.5–5.0) with normal renal profile and acid-base status. He had a normal full blood count, C-reactive protein, plasma glucose, liver function tests, adjusted calcium of 2.32 mmol/L (RI: 2.15–2.55), magnesium 0.73 mmol/L (RI: 0.7–1.0), low serum phosphate 0.56 mmol/L (RI: 0.8–1.4) and raised CK 546 IU/L (RI: 29–195). Spot urine K was 12 mmol/L suggesting appropriate renal response to hypokalaemia. Urine toxicology screen and myoglobin were negative. An electrocardiogram showed sinus tachycardia and marked ST depression in leads II, III, aVF and V4-V6. His chest X-ray and CT brain were normal.
Summary of laboratory parameters in our patient.
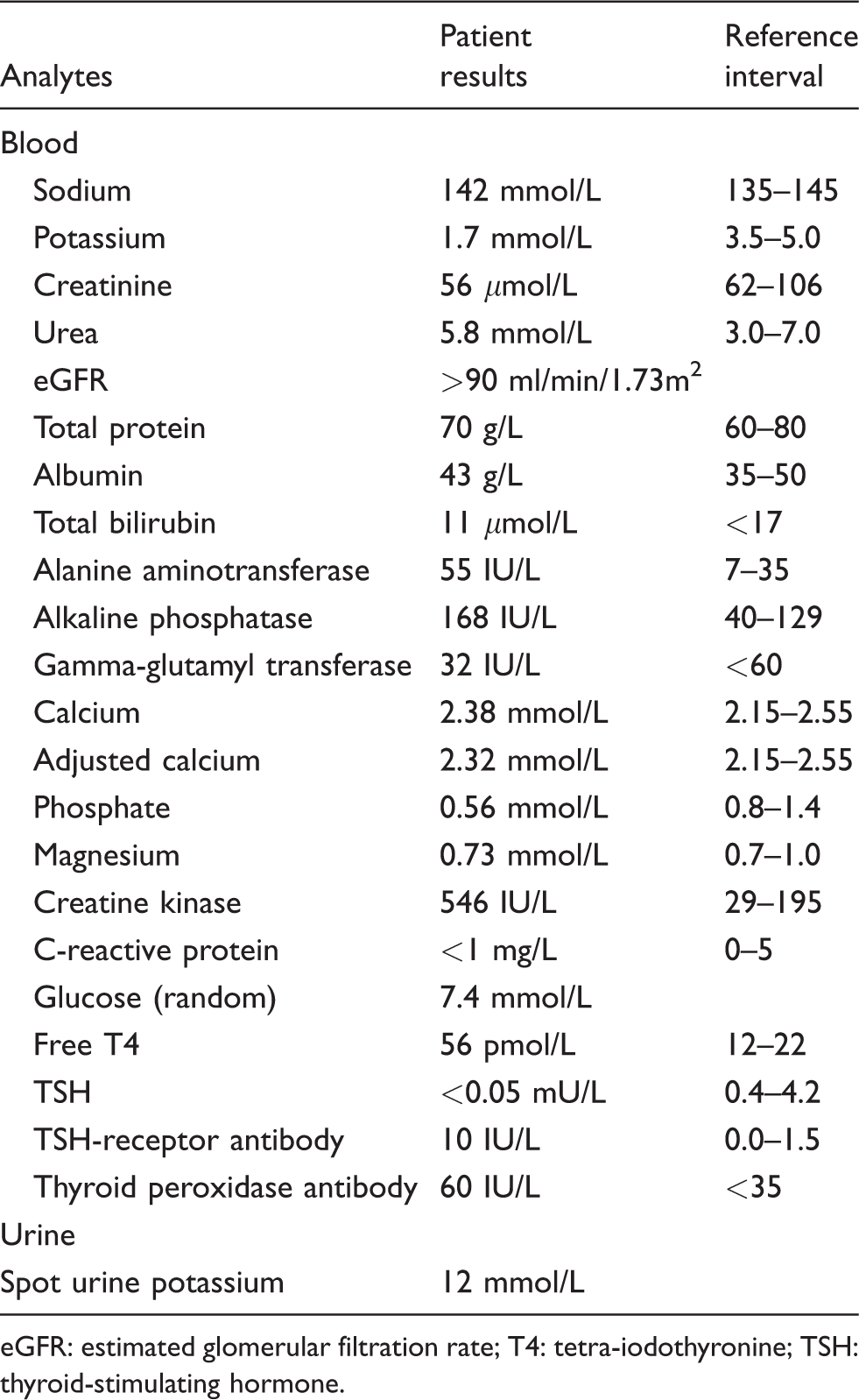
eGFR: estimated glomerular filtration rate; T4: tetra-iodothyronine; TSH: thyroid-stimulating hormone.
In view of his history and laboratory investigations, an initial diagnosis of hypokalaemic periodic paralysis was made. He was treated with intravenous potassium infusion and responded very well to treatment. Serial measurements of serum potassium showed rapid improvement and normalization of his serum potassium level. In view of his symptoms, thyroid function tests were requested. These showed raised serum-free thyroxine of 56 pmol/L (RI: 12–22) with suppressed thyroid-stimulating hormone (TSH) <0.05 mU/L (RI: 0.4–4.2) and raised thyrotropin receptor autoantibodies, consistent with thyrotoxic periodic paralysis (TPP) secondary to Graves’ disease. The patient was commenced on carbimazole 10 mg twice a day. He was closely monitored and carbimazole was discontinued after 18 months of treatment when he became clinically and biochemically euthyroid. Subsequently, he relapsed and had definitive treatment with radioactive iodine to avoid recurrence.
As recent data suggest a link between TPP and genetic mutations in the ion channels for potassium, sodium and calcium, we investigated our patient for a possible underlying genetic defect. The patient consented to genetic analysis which was performed by Centogene AG (Rostock, Germany). The CACNA1S, KCNJ18 and SCN4A genes were analysed by polymerase chain reaction and sequencing of both DNA strands of the entire coding region and the highly conserved exon-intron splice junctions using the reference sequences NM_000069.2, NM_001194958 and NM_000334.4, respectively, for each gene.
He was found to have a heterozygous variant in exon 3 of the KCNJ18 gene (c.576G>C, p.Gln192His), a further heterozygous variant in exon 13 of the SCN4A gene (c.2341G>A, p.Val781Ile) and a third heterozygous variant in exon 12 of the CACNA1S gene (c.1817G>A, p.Ser606Asn). All these variants are reported to be common in normal populations with prevalences of 50%, 2–3% and 1%, respectively (Table 2). Moreover, they are not regarded as disease causing and are unlikely therefore to explain the clinical phenotype of our patient.
Summary of findings of genetic study in our patient.

Discussion
TPP is a rare sporadic muscular disorder characterized by recurrent episodes of generalized muscle weakness associated with hypokalaemia and thyrotoxicosis. Unlike other forms of thyroid disease which are more common in females, TPP mainly affects males of Asian ethnicity (especially from China and Japan) in the second to fourth decades of life. 1
Although the incidence of hypokalaemic periodic paralysis complicating thyrotoxicosis is much lower in Caucasians (0.1%) compared with Asians (2%), the number of non-Asian cases reported has increased recently 2 suggesting TPP may be underdiagnosed in this group. It may occur in association with any of the causes of hyperthyroidism, but Graves’ disease is an underlying cause in most cases.2–5 In TPP, the paralysis results from hypokalaemia due to a rapid and massive transcellular shift of potassium from extracellular to intracellular compartments rather than net potassium loss.6,7 In addition to hypokalaemia, mild to moderate hypophosphataemia may also occur secondary to intracellular shift due to increase sodium-potassium adenosine triphosphate (Na/K-ATPase) pump activity. Serum phosphate levels return to normal without supplementation. The clinical presentation is indistinguishable from familial hypokalaemic periodic paralysis (FHPP), a genetically inherited autosomal-dominant disorder, except that it is accompanied by thyrotoxicosis.
Due to phenotypic similarities with FHPP, it has been proposed that patients with TPP have a skeletal muscle channelopathy 8 and increased susceptibility for enhanced Na/K-ATPase pump activity in response to various hormonal and other triggers. 9 The mechanism by which hyperthyroidism causes hypokalaemic periodic paralysis is complex and whether there is a true genetic predisposition is likewise unclear. Thyroid hormone-responsive elements are present in the promoter region of genes encoding for various Na/K-ATPase (α1, α2, β1, β2 and β-4) subunits expressed in skeletal muscle.10,11 Thyroid hormone has been shown to increase the expression and activity of the Na/K-ATPase pump either directly 11 or indirectly by inducing increased tissue responsiveness to beta adrenergic stimulation, 12 insulin 13 or exercise 14 resulting in rapid potassium influx into the muscle cells, causing hypokalaemia and episodic paralysis. The predilection for TPP in men probably reflects the stimulatory effect of androgens on Na/K-ATPase pump activity, with oestrogens having the opposite effect. Yao et al. showed that episodes of paralysis in Chinese men with TPP were associated with elevated serum testosterone concentrations. 15
To determine whether these predispositions are genetically determined and unmasked by thyrotoxicosis, a number of different candidate genes have been studied. Mutations in voltage-gated calcium 16 (CACNA1S), sodium 17 (SCN4A) and potassium 18 (KCNE3) channel genes have been described. This suggests that TPP may have a genetic basis but is unmasked by thyrotoxicosis. A genetic mutation in KCNJ18 that encodes the inwardly rectifying potassium channel Kir2.6 has also been implicated but was only found in 33% of patients with TPP. 19 The Kir2.6 channel is primarily expressed in skeletal muscle and plays an important role in maintaining stability of the membrane potential. It is transcriptionally regulated by thyroid hormone which means that affected individuals may have a genetic predisposition for paralysis during episodes of thyrotoxicosis. To date, six mutations of the KCNJ18 gene encoding for this channel have been described in TPP patients, 19 where it is associated with an increased prevalence of paralytic episodes. However, the diagnosis of TPP is still primarily based on clinical presentation and presence of hypokalaemia with raised serum free T4, free T3 and suppressed TSH.
Although we were unable to demonstrate previously known mutations as a cause of TPP in our patient, this case highlighted two very important aspects. Firstly, clinical and biochemical findings remain the cornerstone for the diagnosis of TPP. Moreover, since our patient’s clinical phenotype is typical of TPP, it is possible that these or other reported single-nucleotide polymorphisms may in combination increase the susceptibility for TPP. It is also possible that it is a polygenic disease, thus warranting further research to discover putative genetic mutations. It may also be the case that there is no underlying genetic predisposition in many affected patients. Therefore, ultimately, it may be that environmental co-factors (such as exercise, beta adrenergic stimulation, insulin and androgen status) increase the activity of the Na/K-ATPase pump and together or separately play the lead role in triggering severe hypokalaemia in TPP.
Footnotes
Acknowledgements
The authors would like to thank Dr Zafer Yueksel (Centogene AG Rostock, Germany) for his expert opinion on the genetic result.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Written informed consent was obtained from the patient for publication of this case report.
Guarantor
GB.
Contributorship
ER wrote the paper. All authors reviewed and edited the manuscript and approved the final version of the manuscript.