
Abstract
As pathology services become more centralized and automated, the measurement of therapeutic antimicrobial drugs concentrations is increasingly performed in clinical biochemistry or ‘blood science’ laboratories. This review outlines key groups of antimicrobial agents: aminoglycosides, glycopeptides, antifungal agents and antituberculosis agents, their role in managing infectious diseases, and the reasons why serum concentration measurement is important.
Introduction
With the gathering pace of pathology modernization, it is becoming increasingly likely that the measurement of therapeutic antimicrobial drug concentrations is performed in clinical biochemistry or ‘blood science’ laboratories. While the individuals in these setting have vast experience in therapeutic drug monitoring (TDM), this review aims to explain the rationale behind the measurement of key antimicrobial agents. Therefore, the methods and assays used will not be covered as the intended audience will already be very familiar with this topic and other resources are available.
When analysed in UK laboratories, the measurement of therapeutic drug concentrations is subject to the same high standards of quality assurance as other assays. For example, laboratories are required to ensure that assays are appropriately calibrated and quality controlled with internal standards. In addition, external quality assessment for many analytes is available from schemes such as the United Kingdom National External Quality Assessment Service (UK NEQAS) and the Wales External Quality Assessment Scheme (WEQAS).
Antimicrobial agents are measured largely for three reasons: (1) to ensure that sufficient antimicrobial concentrations are being achieved in order to achieve target attainment and hence optimum therapeutic efficacy, (2) to minimize the risks of toxicity and (3) in some cases, to ensure compliance. This review will take key groups of antimicrobials, and explain their role in infectious diseases and why their measurement is necessary.
Aminoglycosides
This group of antimicrobials are broad spectrum and are usually used to treat serious infections. 1 They are used most frequently to treat infections caused by aerobic Gram-negative bacilli. Some are used in combination with other antimicrobials to treat infections caused by Gram-positive organisms, such as endocarditis. 2 In addition, some have activity against mycobacteria. They are usually inactive against anaerobic organisms. Examples of commonly used (and therefore measured) aminoglycosides, include gentamicin and amikacin.
These large molecules consist of one or several aminated sugars joined in glycosidic linkages to a dibasic cyclitol. In most clinically used aminoglycosides, the latter is 2-deoxystreptamine and it is streptidine in streptomycin and derivatives and fortamine in the fortimicin series. 3 Entry of these polar molecules into the bacterial cell is via a self-promoted uptake process across the bacterial outer membrane that involves drug-induced disruption of Mg2+ bridges between adjacent polysaccharide molecules.4,5 Transport across the cytoplasmic membrane is via an electron transport process termed an energy-dependant phase I (EDP-I) mechanism. 6 The aminoglycoside then binds to the 30 S unit of the bacterial ribosome via another energy-dependant phase process (EDP-II). 6 Rather than inhibiting the formation of the peptide initiation complex, the drug disrupts the elongation of the peptide chain by impairing proof-reading and therefore results in translational inaccuracy. 7 The faulty proteins may be inserted into the bacterial membrane and lead to increased uptake of the drug. 8
Aminoglycosides are water soluble and are therefore largely administered intravenously, although intramuscular administration may occur for some drugs. In addition to their high level of solubility in water, they exhibit a low level of protein binding (approximately 10%), and as such are widely distributed in the vasculature and in most soft tissues. 9 Their bactericidal activity is concentration dependent, and plasma concentrations approximately equate to the concentration of drug in interstitial fluids following repeat dosing and the achievement of steady-state. 10 A higher volume of distribution is seen in patients with increased third space volume, such as those with ascites and burns, while a lower volume of distribution is seen in patients with obesity. 11
There are exceptions to the effective distribution of aminoglycosides around the body. Due to their large size, polycationic charge and lipid insolubility, they do not easily cross membranes. Except in the new born, they demonstrate poor penetration across blood–cerebrospinal fluid (CSF) and blood–brain barriers. 12 Exceptions to this include the renal tubular cells and inner ear. 13 Aminoglycosides are concentrated in the renal proximal tubules, resulting in increased concentrations compared with plasma. 13 For this reason, gentamicin is often used in the treatment of urosepsis.
All aminoglycosides have the ability to cause nephrotoxicity, ototoxicity and neuromuscular block. Their toxicity is directly related to their positive electrical charge at physiological pH. Parenteral aminoglycosides are almost entirely excreted, unaltered, by the kidney through glomerular filtration. This route of excretion is the basis of their nephrotoxicity, although the full mechanism is not understood. It is thought to be related to their uptake in the epithelial cells of the proximal renal tubules 14 following glomerular filtration. 15 Once accumulated in these cells, they produce a wide range of morphological and functional effects, resulting in non-oliguric renal failure, with a slow rise in serum creatinine and a hypo-osmolar urinary output. 16 Less commonly, acute oliguric renal failure may occur.
The reported incidence of nephrotoxicity varies according to study and the definitions used, with most reports being in the range of 5 to 25%. 1 Aminoglycoside-related nephrotoxicity is related to length of administration and dose. Dosing regimens with extended intervals have been shown to be associated with less nephrotoxicity, 16 and are being increasingly used. Many clinicians use a once-daily dose of 5 to 7 mg/kg, based on ideal body weight. However, extended-interval dosing is not suitable for all patient-groups (e.g. those with severe burns or gross ascites), and current national guidance suggests various dosing regimens for gentamicin. 17 A survey in the USA as far back as 2000 found that 75% of centres had adopted extended-interval dosing for several reasons including ease, reduced toxicity and increased efficacy. 18 Amikacin is often dosed as once daily at 15 mg/kg, based on ideal weight with a maximum daily dose of 1.5 g.
Ototoxicity is manifested by cochlear or vestibular damage, and is normally irreversible and may also occur after cessation of treatment. Toxicity results in the destruction of the sensory hair cells in the cochlea and vestibular labyrinth, and repeated exposure to an aminoglycoside increases the risk of this side-effect. 19 The incidence of aminoglycoside-related ototoxicity varies by study. This is in part due to the methods used to detect hearing loss. Aminoglycoside use causes a loss in perception of high-frequency sounds initially. The inability to hear these wavelengths does not affect the ability to recognize speech – lower frequencies are important for this. Therefore, some studies that use high-frequency audiograms to detect hearing loss may report high instances of ototoxicity even if perceptible hearing loss is not reported. 19 Treatment courses of aminoglycosides should be kept as short as clinically possible and serial high-frequency audiograms should be considered for courses exceeding four days. 1
Using any dosing regimen, it is critical to measure serum concentrations of aminoglycosides, particularly in prolonged courses. With multiple daily dose regimens, both trough and peak serum concentrations must be monitored, usually starting with the third dose. A predose (trough) serum concentration should be taken immediately before the dose is due. In order to avoid toxicity, this serum concentration of gentamicin must be less than 1 mg/L before a subsequent dose is given. Trough serum amikacin concentrations should be below 5 mg/L. If the serum concentration exceeds this, then the dose must be withheld and the serum concentration measured again in 12 h. Peak serum concentrations should be measured to ensure that therapeutic concentrations are being obtained. This should be measured 60 min after the beginning of the infusion (which usually takes 30 to 45 min). If satisfactory concentrations are obtained, and renal function remains stable, concentrations may be measured twice weekly in most cases.
One of the advantages of extended-interval dosing is the applicability of nomograms to guide the dosing; the Urban-Craig and Hartford nomograms are applicable to the 5 and 7 mg/kg gentamicin regimens, respectively (Figures 1 and 2).20,21 These nomograms allow for considerable flexibility in the timing of serum concentrations, and only one concentration is required (e.g. with the Hartford nomogram, a concentration is taken 6–14 h postdose). The position of the concentration on the nomogram determines the dosing interval, the dose itself remaining the same. For example, if the concentration falls below the lowest line on the Hartford nomogram, a dosing regimen of 7 mg/kg every 24 h is followed; if it falls between the lowest and middle lines, the same dose is given every 36 h, etc.). Clearly, the timing of the concentration relative to the dose is critical in interpretation.
Urban-Craig nomogram.
20
Hartford nomogram.
21
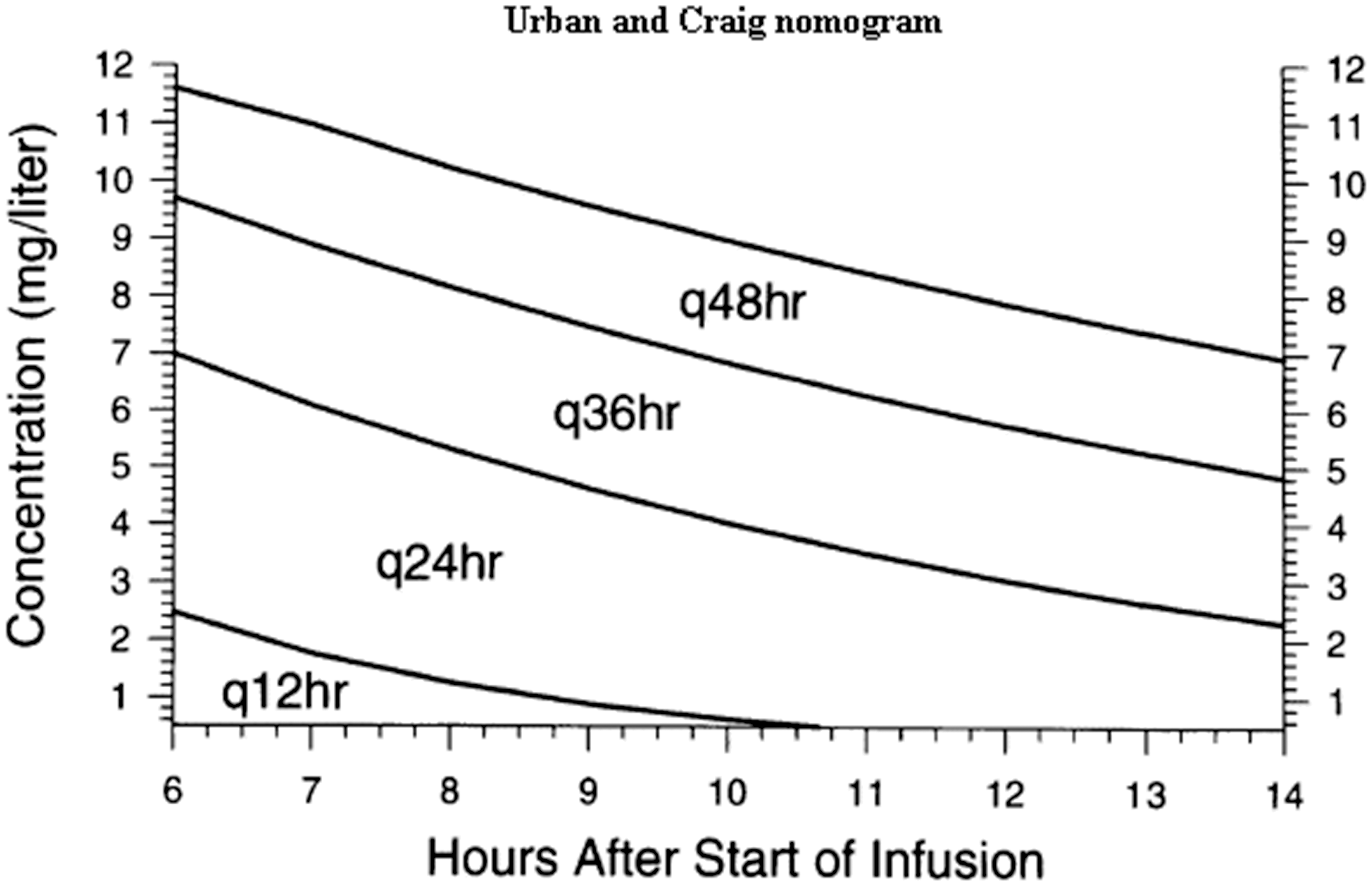
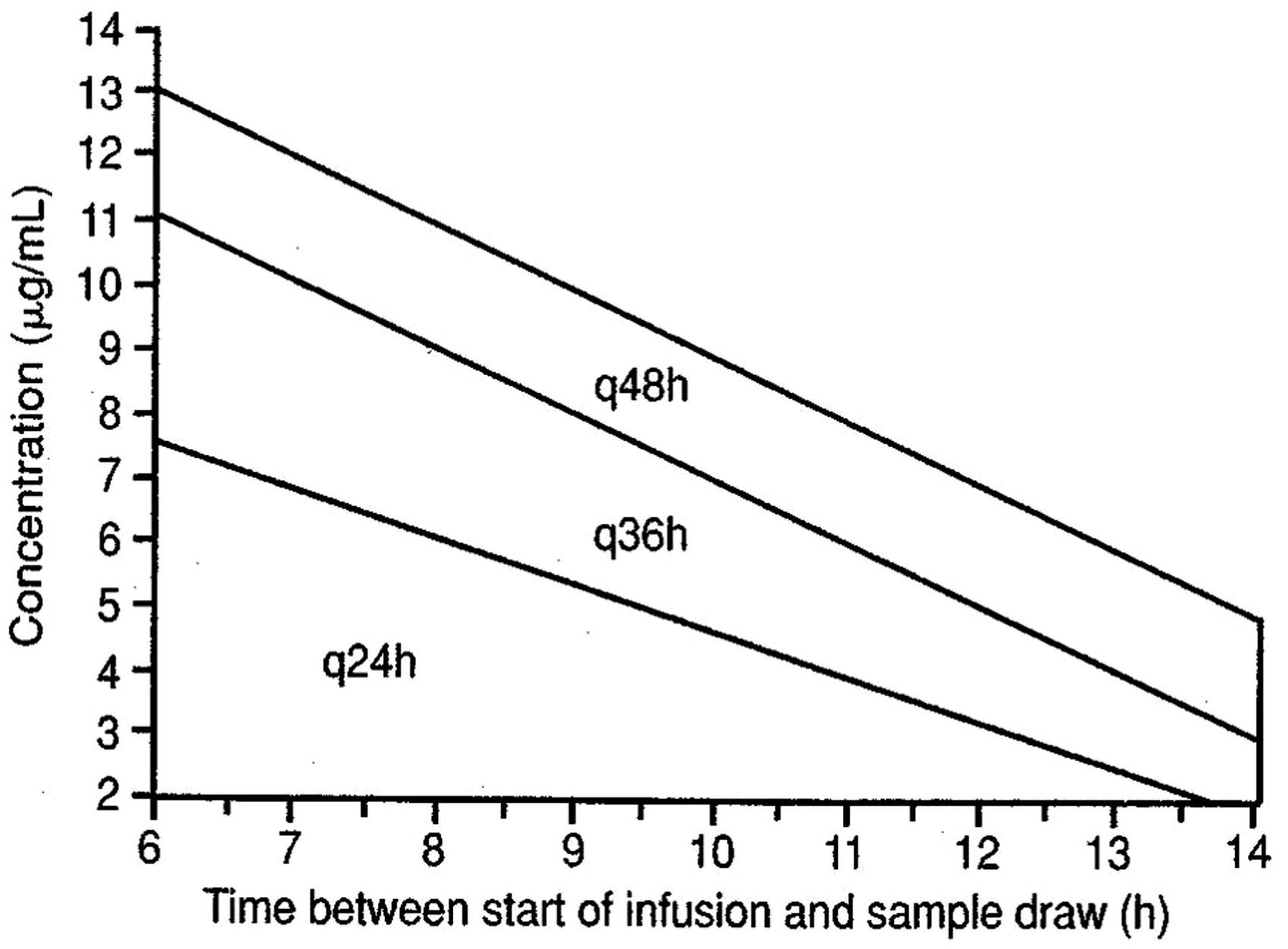
Stat doses are often prescribed in surgical prophylaxis and sepsis. In such cases, the estimation of renal function and measurement of therapeutic concentrations are usually not required unless subsequent doses are to be administered.
Glycopeptides
These antibiotics are active against aerobic and anaerobic Gram-positive organisms. Members of this class currently licensed in UK are teicoplanin and vancomycin.
Vancomycin has a complex tricyclic glycopeptide structure with a heptapeptide skeleton. It has a relatively high molecular weight of 1485.73 daltons. Teicoplanin is a mixture of several related glycopeptide analogues and has a molecular weight of around 1900 daltons. In the circulation, more than 90% of it is protein bound.
Vancomycin and teicoplanin exert their bactericidal activity by inhibiting peptidoglycan biosynthesis in the bacterial cell wall. They form a stoichiometric 1:1 complex with the peptidoglycan precursor UDP-N-acetylmuramylpentapeptide, by forming hydrogen bonds, thereby inhibiting the transglycosylase enzyme involved in adding the peptidoglycan precursor to the growing peptidoglycan chain, probably by steric hindrance. Other enzymes involved in cross-linkage of adjacent peptidoglycan chains are also inhibited. 22
Glycopeptides are administered intravenously for systemic infection. Oral absorption is poor, and this route of administration is only used to treat Clostridium difficile infection. When administered intravenously, glycopeptides distribute well into most body fluids with the exceptions of CSF and aqueous humour. Intraperitoneal administration is used to treat peritonitis associated with continual ambulatory peritoneal dialysis (CAPD peritonitis). Treatment of meningitis and endophthalmitis with vancomycin requires intraventricular and intravitreal administration respectively. Both teicoplanin and vancomycin are excreted by renal elimination, mostly by glomerular filtration.
Vancomycin and teicoplanin exhibit time-dependent bactericidal activity; no significant change in activity is obtained by altering concentrations above the minimum inhibitory concentration (MIC) of the organism. Outcome correlates best with the area under the concentration-time curve (AUC)/MIC ratio as shown below (Figure 3).
23
Concentration/time curve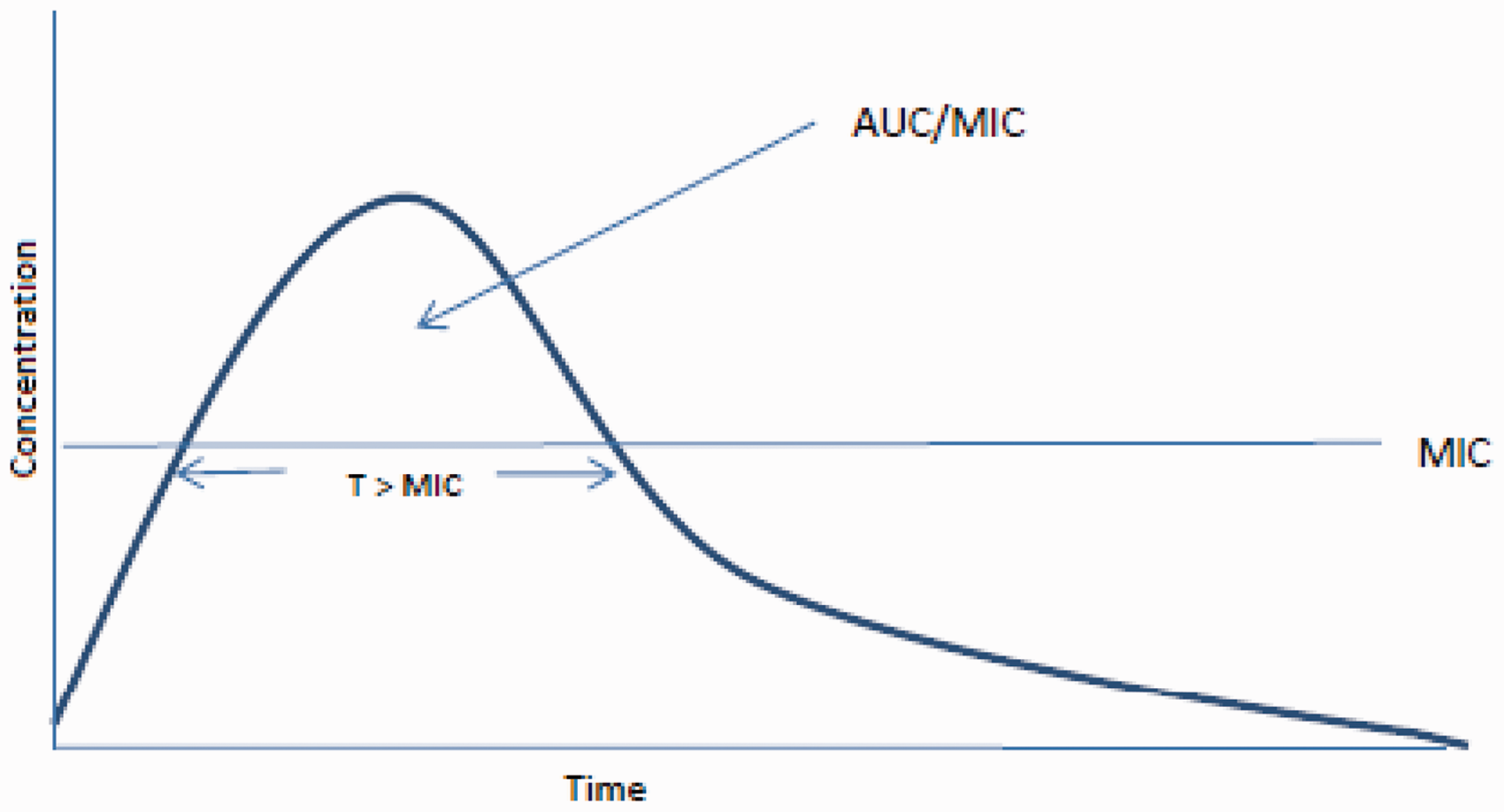
Intravenous dosage is usually by twice daily infusion; the frequency of administration and dose may need reduction in renal impairment. Some studies have suggested better attainment of pharmacokinetic/pharmacodynamic targets with continuous 24 h infusion regimens, and these regimens are being increasingly used. 24 Intravenous regimens of teicoplanin comprise three loading doses at 12-h intervals, followed by a maintenance once daily schedule. The maintenance regimen may need adjustment in renal impairment.
Although there does appear to be some risk of nephrotoxicity associated with vancomycin use, data are conflicting as to the true extent of this problem. Earlier reports, when preparations of vancomycin contained impurities, likely over-estimated this risk, and the presence of other factors that could impair renal function often makes more recent studies difficult to interpret. An association between high trough vancomycin concentrations and nephrotoxicity has been seen in some studies. 25
Vancomycin-related ototoxicity is unusual. However, a recent study has demonstrated high-frequency hearing loss detectable by audiometry in 19% of patients over the age of 53 years treated with vancomycin. 26 A relationship between ototoxicity and serum vancomycin concentrations has not been established.
These adverse effects are less common with teicoplanin than they are with vancomycin. Dose-related nephrotoxicity with teicoplanin has, however, been demonstrated in animal studies, and a slight increase in auditory threshold has been noted, albeit rarely, in patients treated with teicoplanin.27,28
The primary role of monitoring serum vancomycin concentration is to ensure that an adequate concentration of drug is maintained in order to achieve the pharmacokinetic and pharmacodynamic targets that correlate with optimum efficacy (i.e. AUC/MIC > 400). In intermittent dosage regimens, trough serum concentrations are monitored, starting with the third dose, with the aim of maintaining a concentration of 10 to 15 mg/L for most infections. For complicated infections or less sensitive strains (i.e. strains with a higher MIC), a concentration of 15 to 20 mg/L is necessary. 17 High peak serum concentrations correlate with neither efficacy nor toxicity, and measurement is therefore unnecessary. In continuous infusion regimens, steady-state serum concentrations are monitored, with the aim of maintaining a concentration of 15 mg/L.
The good safety profile of teicoplanin means that serum concentration monitoring is usually not necessary. Trough serum teicoplanin concentrations are usually monitored only in severe or particularly difficult infections, such as endocarditis and osteomyelitis, with the aim of maintaining the trough concentration above 20 mg/L. Peak serum teicoplanin concentrations are not measured as they correlate with neither efficacy not toxicity.
Antifungal agents
Azoles
The azoles that routinely require TDM are the triazoles itraconazole, voriconazole and posaconazole. 29 These drugs exhibit concentration-independent killing, the most important correlate of treatment efficacy being the AUC/MIC ratio. 30
Antimicrobial activity
Itraconazole and posaconazole contain a piperazine-phenyl-triazole side chain. Itraconazole has broad spectrum activity and it is used as both prophylaxis and treatment in a wide variety of clinical settings and fungal infections. Itraconazole is active against Candida spp., Cryptococcus spp. and Aspergillus spp., and is used in the treatment of histoplasmosis, coccidiomycosis, sporotrichosis, dermatophyte infections and penicilliosis. Itraconazole also has some activity against zygomycetes. Posaconazole contains a furan ring with fluorine substituted for chlorine. The spectrum of activity of posaconazole is similar to that of itraconazole, its main advantage being against filamentous fungi, particularly Zygomycetes, which demonstrate lower MICs to posaconazole. 31
Voriconazole is a synthetic triazole derivative in which one triazole ring is replaced with a fluorinated pyrimidine and an α-methyl group is added to the propanol backbone. It has broad spectrum antifungal activity, being active against most endemic dimorphic fungi, yeasts (including fluconazole-resistant Candida sp.), moulds and filamentous fungi. Voriconazole is used as first-line therapy for invasive candidiasis caused by species with reduced susceptibility to fluconazole, invasive aspergillosis and serious infections caused by Scedosporium or Fusarium spp. Voriconazole has no significant activity against zygomycetes.
The mode of action of the triazoles is inhibition of fungal cytochrome P450-dependent C-14-α sterol demethylase. This interferes with ergosterol biosynthesis resulting in increased permeability of the fungal cell membrane.
Dosage and pharmacokinetics
Itraconazole can be administered either orally, using a once or twice daily regimen or intravenously. Intravenous regimens comprise a loading phase of two days followed by a once-daily maintenance phase. The oral bioavailability of itraconazole is highly variable; absorption of the oral solution is better than that of capsules, and is improved by fasting.
Voriconazole also has oral and intravenous formulations. Both regimens comprise a twice daily loading phase of 24 h followed by a twice daily maintenance regimen. Oral absorption is best in the fasting state. The intravenous formulation comprises voriconazole complexed with sulfobutyl-ether-β-cyclodextrin (SEBCD).
Posaconazole is currently available in tablet and liquid oral formulations and an intravenous formulation is likely to be marketed soon. It is administered two to four times daily. It may take up to 10 days to achieve steady-state concentrations and loading regimens cannot be applied because absorption is saturable. Absorption of posaconazole is improved by food intake, and is best after a meal high in fat content; it is reduced by agents that reduce gastric acidity. In addition, there is considerable interpatient variability in absorption, an important consideration being significant impairment in patients with mucositis. 32
Itraconazole exhibits non-linear pharmacokinetics and does not, therefore, have a half-life. It distributes well into tissues, and has a large volume of distribution. 33 Penetration into CSF and vitreous are poor, partly due to very high protein-binding.
Voriconazole exhibits non-linear pharmacokinetics, which reflects saturable clearance mechanisms. An important consequence of this is unpredictable variation in serum voriconazole concentration for given dose changes. Tissue distribution, including ocular and central nervous system penetration, is excellent, and voriconazole is the drug of choice for aspergillosis affecting the central nervous system. Voriconazole does not, however, achieve good concentrations in urine. 34
More than 98% of circulating posaconazole is protein bound. It has a long half-life of 25 to 31 h and a large apparent volume-of-distribution of 343 to 1341L.35,36 CSF penetration is poor, and ocular penetration inferior to that of voriconazole. Urinary excretion is minimal, although good concentrations of the drug are achieved in the renal parenchyma.
Itraconazole is metabolized via oxidative mechanisms, predominantly via the CYP3A4 isoenzyme, into several metabolites. Hydroxy-itraconazole, one of these metabolites, has similar activity to itraconazole. This produces a disparity between bioassay and high-performance liquid chromatography (HPLC) methodology when used to assay serum itraconazole concentrations; bioassay over-estimating the concentration by as much as five-fold. 37
Voriconazole is metabolized via oxidative mechanisms by several cytochrome P450 isoenzymes (predominantly CYP3A4, CYP2C9 and CYP2C19). 38 Polymorphisms in CYPC19 result in considerable variation in clearance. Co-administration of inducers of the cytochrome P450 system, such as rifampicin, phenytoin and carbamazepine, can significantly reduce serum voriconazole concentrations. In addition to being a substrate, voriconazole is also an inhibitor of these isoenzymes, resulting in increased exposure to several other agents (e.g. cyclosporine and several benzodiazepines).
Posaconazole is largely excreted unchanged in faeces. A small proportion is metabolized into glucuronide conjugates by the CYP450 isoenzymes. 36
Adverse effects
Itraconazole is well tolerated. Adverse effects are rare, the most common being a rise in serum bilirubin and aminotransferases, which affect about one-third of patients. Other less common adverse effects include neurological and gastrointestinal disturbances. 39 These adverse effects occur more frequently at higher concentrations.
Visual disturbances (photopsia) occur in about one-third of patients treated with voriconazole. Although common, these disturbances are transient. Less common side-effects include cutaneous photosensitivity, altered liver function (usually cholestasis) and neurological symptoms, commonly a reversible encephalopathy. The occurrence of photopsia, hepatotoxicity and encephalopathy is related to increasing serum voriconazole concentrations.40,41
Posaconazole is generally well tolerated. The most common adverse effects are gastrointestinal symptoms, rash and abnormal liver function tests.
Serum concentration monitoring
Serum itraconazole concentrations are useful to guide management in both prophylaxis and treatment of invasive fungal infection. These infections breakthrough more commonly in neutropenic patients with trough itraconazole concentrations of <0.25 to 0.5 mg/L, and clinical outcomes with a variety of invasive fungal infections, including aspergillosis, are better with higher trough concentrations. Toxicity is also concentration related. The British Society for Medical Mycology recommends that a trough serum itraconazole concentration is determined five to seven days after starting treatment or dose adjustment, and that monitoring is repeated at regular intervals. A target trough concentration of 0.5 to 1 mg/L is recommended for both prophylaxis and therapy when serum concentrations are measured by either HPLC or mass spectrometry. 29
Serum voriconazole concentrations correlate well with both clinical efficacy as well as with toxicity. As a result of the pharmacokinetic variability of voriconazole, they are also subject to wide (≈100-fold) inter-patient variation. In order to optimize dosage, TDM should therefore be performed in most patients being treated with voriconazole. 41 The British Society for Medical Mycology recommends that a trough serum voriconazole concentration is determined within the first seven days after starting treatment or dose adjustment, and that monitoring is repeated at regular intervals. It is important that sampling is repeated because serum voriconazole concentrations may progressively accumulate in patients in whom clearance is saturated. A target trough concentration of >1 mg/L is recommended for both prophylaxis and therapy. 29 A higher target (e.g. 2 mg/L) should be applied in disease with a particularly poor prognosis (e.g. infection within the central nervous system or multifocal infection). It has been shown that maintaining serum voriconazole concentrations <5.5 mg/L prevents attributable toxicity. 42
The pharmacokinetic and pharmacodynamic variability of posaconazole, together with the relationship between serum concentration and clinical efficacy, makes the monitoring of serum concentrations an important consideration in patients treated with posaconazole. The British Society of Medical Mycology makes different recommendations for patients receiving posaconazole for prophylaxis and treatment. 29 In prophylaxis, patients should have serum trough itraconazole concentrations determined seven days after initiation, and a target concentration of >0.7 mg/L is applied. A trough concentration can also be measured 48 h after initiation, but a lower target of 0.35 mg/L is then applied. In treatment of established infection, a trough serum concentration should be determined seven days after initiation, and maintained above 1 mg/L. Serum trough concentrations should be repeated regularly, and seven days following dose change.
Flucytosine
Flucytosine is a fluorinated pyrimidine analog with a relatively low molecular weight of 120 daltons. Most medically relevant yeasts, including Candida spp. and Cryptococcus spp., are susceptible to flucytosine. However, resistance to this agent develops rapidly, and it therefore cannot be used as monotherapy. The main clinical application of flucytosine is as combination therapy for cryptococcal meningitis and candida infections in sites into which antibiotic penetration is poor. Flucytosine also has activity against Aspergillus spp. and rare dematiceous fungi that cause chromoblastomycosis. 43
Flucytosine is transported into fungi by membrane permeases. It is then deaminated into 5-fluorouracil, which is further metabolized into several derivatives which interfere with DNA synthesis through inhibition of thymidylate synthetase and nucleotide depletion.
Both oral and intravenous formulations of flucytosine are available. The same six to eight hourly dosage regimen is applied for both formulations. Oral bioavailability is excellent, in the region of 90%. 44 Flucytosine is widely distributed into tissues and body fluids, including CSF, eye compartments and urine. 44 There is, however, significant inter-patient variability in serum concentrations. Flucytosine is largely excreted unchanged by glomerular filtration, and renal impairment can therefore produce elevated serum flucytosine concentrations.
The best correlate of clinical efficacy of flucytosine is the fraction of the dosing interval for which serum concentration exceeds MIC. 45
The most important adverse effect of flucytosine is myelotoxicity. This adverse effect is dose-related, toxicity being associated with sustained elevated flucytosine concentrations (>100 mg/L). 46 Leucopenia usually resolves if the flucytosine dosage is promptly reduced. Transient hepatotoxicity can also result from persistently elevated concentrations.
TDM is essential in patients being treated with flucytosine, largely to minimize the risks of myelotoxicity. Additional benefits of monitoring serum flucytosine concentrations are the minimization of the risks of the emergence of resistance and maximization of clinical efficacy. The British Society for Medical Mycology recommends that serum concentrations are measured within 72 h of treatment initiation. A trough flucytosine concentration of 20 to 40 mg/L and a peak concentration of no greater than 100 mg/L are recommended. The short half-life of flucytosine means that serum concentrations can change quickly, and regular measurement is therefore necessary.
Antituberculosis therapy
Tuberculosis (TB) is an infectious disease caused by the bacterium Mycobacterium tuberculosis. TB is primarily a respiratory disease, although it can affect any part of the body and it is spread from person-to-person via expectorated droplets by individuals with active pulmonary disease. TB is a significant international problem and is the second only to HIV/AIDS with regard to deaths caused globally by a single infectious agent. The World Health Organization estimates that in 2012, 8.6 million people fell ill with TB and 1.3 million died from TB, the vast majority of these occurring in Asia and sub-Saharan Africa. 47
This global epidemic is further complicated by the increase in drug resistant M. tuberculosis. Multidrug-resistant tuberculosis (MDR-TB) is a form of TB caused by bacteria that do not respond to, at least, isoniazid and rifampicin, the two most effective, first-line anti-TB drugs. There were approximately 450,000 cases of MDR-TB globally in 2012. More than half of these cases were in India, China, the Russian Federation and Africa. 47
TB in the UK is treated with a prolonged combination of antibiotics as per national guidance. Standard treatment of pulmonary TB consists of an initial four-drug regimen for two months (isoniazid, rifampicin, pyrazinamide and ethambutol), followed by a continuation phase of four months with isoniazid and rifampicin. 48
The measurement of antituberculosis drugs may be performed if poor absorbance or compliance is suspected. Concentrations may also be measured when a patient is not responding as expected. Finally, many of the drugs used, particularly second- and third-line drugs that are used in drug-resistant TB have significant toxicity profiles.
First-line therapy
TDM is rarely performed routinely in patients with susceptible TB. Limited retrospective cases series have attempted to demonstrate that subtherapeutic drug concentrations are associated with inferior disease resolution.49,50,51 However, shortcomings of these papers include the use of poor controls, small sample size and selection bias. Many of these limitations are a direct result of the infrequent measurement of antituberculosis drugs in clinical practice. TDM is often only performed because a patient is not responding to treatment. One Danish study demonstrated that subtherapeutic concentrations were often noted and that treatment failure occurred more frequently when plasma concentrations of isoniazid and rifampicin were both below the reference ranges. 52 There is also discrepancy between studies in sampling times. Blood drawn at 2 h postdose should provide Cmax concentrations, which can be confirmed by a lower concentration in a sample taken 6 h postdose. Should the concentration taken at 6 h be similar, or higher than the concentration taken at 2 h, then this is indicative of delayed absorption. Two such concentrations are rarely taken for practical reasons.
Cost is often cited as a reason for not performing TDM more frequently. One review estimates that the increased cost associated in measuring drug concentrations on all TB patients in the United States would be more than cancelled out by the savings gained by the reduced need to prolong TB treatment in 10% of their cohort. This does, however, assume that all patients who require prolonged TB treatment do so because of subtherapeutic drug concentrations. The review does concede that larger, prospective studies are required to establish the precise correlation between plasma drug concentrations and disease outcome. 53
Summary of first-line antituberculosis treatment.

Adapted from BTS. 54
Summary of second- and third-line antituberculosis treatment.

Adapted from BTS. 54
Summary
Monitoring serum concentrations is therefore an integral part of treatment with several antimicrobial agents. It should, however, be remembered that serum concentrations are only a surrogate marker in many situations. This is particularly true when the site of infection is difficult to penetrate with antibiotics. For example, serum antibiotic concentrations will often bear little relation to antibiotic concentrations in CSF, epithelial lining fluid and bone. In addition, the best predictors of antibiotic efficacy are frequently more complex pharmacokinetic parameters, such as AUC/MIC. In these situations, while serum concentration monitoring offers a relatively convenient guide, efficacy and toxicity will be determined by a complex interplay of several other factors.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Guarantor
RJS.
Contributorship
Both authors contributed equally to writing of the review manuscript.