
Abstract
Background
Capric acid (FA10:0, decanoic acid) is a medium-chain fatty acid abundant in tropical oils such as coconut oil, whereas small amounts are present in milk of goat, cow, and human. Orally ingested FA10:0 is transported to the liver and quickly burnt within it. Only few reports are available for FA10:0 concentrations in human plasma.
Methods
Fasting (n = 5, male/female = 3/2, age 31 ± 9.3 years old) and non-fasting (n = 106, male/female = 44/62, age 21.9 ± 3.2 years old) blood samples were collected from apparently healthy Japanese volunteers. The total FA10:0 in the plasma were measured by high-performance liquid chromatography after derivatization with 2-nitrophenylhydrazine followed by UV detection.
Results
Inter and intra-assay coefficient of variation of FA10:0 assay at three different concentrations ranged in 1.7–3.9 and 1.3–5.4%, respectively, with an analytical recovery of 95.2–104.0%. FA10:0 concentration was below detection limit (0.1 µmol/L) in each fasting human plasma. FA10:0 was not detected in 50 (47.2%) of 106 non-fasting blood samples, while 29 (27.4%) plasma samples contained FA10:0 less than or equal to 0.5 µmol/L (0.4 ± 0.1), and 27 (25.5%) contained it at more than 0.5 µmol/L (0.9 ± 0.3).
Conclusion
A half of the non-fasting plasma samples contained detectable FA10:0. This simple, precise, and accurate high-performance liquid chromatography method might be useful for monitoring plasma FA10:0 during medium-chain triglycerides therapy.
Introduction
Medium-chain triglycerides (MCT) are composed of medium-chain fatty acids (MCFA), mainly octanoic acid and capric acid (decanoic acid, FA10:0). Unique metabolic properties make it as lipid of interest to improve various clinical conditions and have been extensively reviewed.1–6 Beside synthetic MCT, there are some natural source of MCFA. Coconut and palm kernel oils are rich in MCFA with more than 50 wt.% of fatty acids. MCFA are also present in milk from goat, cow, and human, for instance MCFA in bovine milk make up 4–12% of all fatty acids.7,8 Therefore, milk and milk products, specially butter are the important dietary source of FA10:0 in human nutrition. Measurement of plasma concentration of FA10:0 remains challenging due to its trace availability compared to long-chain fatty acids (LCFA) in systemic circulation, but can provide valuable information during therapeutic use of MCT.
Though gas chromatography (GC) is currently the routine procedure for FA analysis, it is associated with several disadvantages. 9 In particular, FA10:0 is relatively volatile than LCFA, and methyl derivatization adds more volatility to FA10:0 possibly resulting in low recovery during preanalytical steps for GC. High-performance liquid chromatography (HPLC) with appropriate derivatization overcomes this problem, further adding the advantages of speed, resolution, sensitivity, and specificity. A wide variety of derivatization and detection systems are available including UV/visible, fluorescence, chemiluminescence, electrochemical, light scattering, and mass spectrometry detection for the HPLC analysis of FA.9,10 One of the simple and reliable HPLC approaches is derivatization of FA to its hydrazide using 2-nitrophenylhydrazine hydrochloride (2-NPH·HCl), followed by reversed phase separation and UV/visible detection.11,12 Enzymatic methods for determination of FA have also been described.13,14 Although a large number of techniques have been described for accurate and reproducible measurement of the FA in the plasma, none of them were designated specifically for FA10:0. Importantly, there is paucity of statistical data on precision and accuracy in the measurement of plasma FA10:0.
We describe here (a) the development of a specific and sensitive HPLC method for the measurement of FA10:0 in the plasma, (b) analytical consideration for the measurement of FA10:0, and (c) plasma FA10:0 concentration in fasting and non-fasting healthy humans.
Materials and methods
Chemicals
2-NPH·HCl, 1 -(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1-EDC·HCl), undecanoic (FA11:0), myristic (FA14:0), palmitic (FA16:0), heptadecanoic (margaric acid, FA17:0), and stearic acid (FA18:0) were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). FA10:0 and pyridine were purchased from Wako Pure Chemical Industry (Osaka, Japan). Methanol and water were of HPLC grade and from Wako Pure Chemical Industry (Osaka, Japan). FA labeling reagents were obtained from YMC Co. (Kyoto, Japan).
Synthesis of standards
We used 2-NPH-labeled FA10:0, FA14:0, FA16:0, and FA18:0 as standards while FA11:0 and FA17:0 as the internal standards (IS). The solution of respective FA (FA10:0, FA11:0, FA14:0, FA16:0, FA17:0, and FA18:0; 5.0 mmol each), 2-NPH·HCL (5.5 mmol), 1-EDC·HCl (5.5 mmol), pyridine (3% v/v) in ethanol was heated at 60℃ for 20 min to synthesize specific 2-NPH-labeled FA (FA-NPH). Formation of the products was monitored by thin layer chromatography (Silica gel 60 F254, Merck Co., Darmstadt, Germany). The reaction products were purified by fractionation after thoroughly washing several times with HCl (5%), neutralized by NaHCO3 (5%), distilled water, saturated solution of NaCl sequentially, and dried over Na2SO4, followed by the evaporation of the organic solvent using a rotary evaporator. Purification of the product by column chromatography on a silica gel 60 N (100–120 µm, Kanto Chemical Co., Inc., Tokyo, Japan) using chloroform–hexane (2:1, v/v) as an eluent and recrystallization of a homogeneous effluent from methanol gave pure FA-NPH as yellowish needle shaped crystals.
The melting points of the synthesized FA-NPH were measured by a micro melting point apparatus (Yanaco New Sciences Inc., Kyoto, Japan) and ranged from 94.1–94.8 (FA10:0-NPH), 96.5–97.3 (FA11:0-NPH), 103.8–104.7 (FA14:0-NPH), 107.5–108.0 (FA16:0-NPH), 109.4–109.8 (FA17:0-NPH), and 110.3–111.0℃ (FA18:0-NPH). The purity of the standards was assessed by 1H and 13C-nuclear magnetic resonance (NMR) using JEOL JNM-AL400 (Tokyo, Japan) with CDCl3 as solvent. 1H-NMR (CDCl3, 400 MHz) of the major rotamer of FA10:0-NPH, δ (ppm) 8.97 (bs, 1H, 8.16 (1H, dd, J = 1.4, 7.2 Hz, H-3), 7.47 (1H, dt, J = 1.4, 7.2 Hz, H-5), 7.04 (1H, dd, J = 0.9, 7.2 Hz, H-6), 6.86 (1H, dt, J = 0.9, 7.2 Hz, H-4), 2.31 (t, J = 7.3 Hz, 2H, –CO–CH2’–), 1.70 (2H, quin, J = 7.2 Hz, H-3’), 1.21–1.35 (m, 12H, –CH2’–), and 0.88 (t, 3H, J = 6.8 Hz, H-10’). 13C NMR (CDCl3, 100 MHz) δ (ppm) 172.94 (C1’), 145.27 (C1), 136.18 (C5), 133.40 (C2), 126.62 (C3), 119.06 (C4), 114.23 (C6), 34.44 (C2’), 31.99 (C8’), 29.56, 29.45, 29.43, 29.39 (C4’ to C7’), 25.53 (C3’), 22.80 (C9’), 14.27 (C10’). Molecular characterizations of synthesized FA-NPH were carried out by an electrospray ionization mass spectrometer (Thermo Scientific Finnegan LXQ, CA, USA). Base peaks in the mass spectra of FA10-NPH (M = 307.2) are formed by ionization of the [M+H]+ ion at m/z 308.2 and [M–H]– ion at m/z 306.3 on the positive and negative-ion mode, respectively (Figure 1).
Electro spray ionization mass spectra of NPH-derivative of capric acid in (a) positive- and (b) negative-ion mode. Key diagnostic ion peaks are annotated in bold type with their m/z value.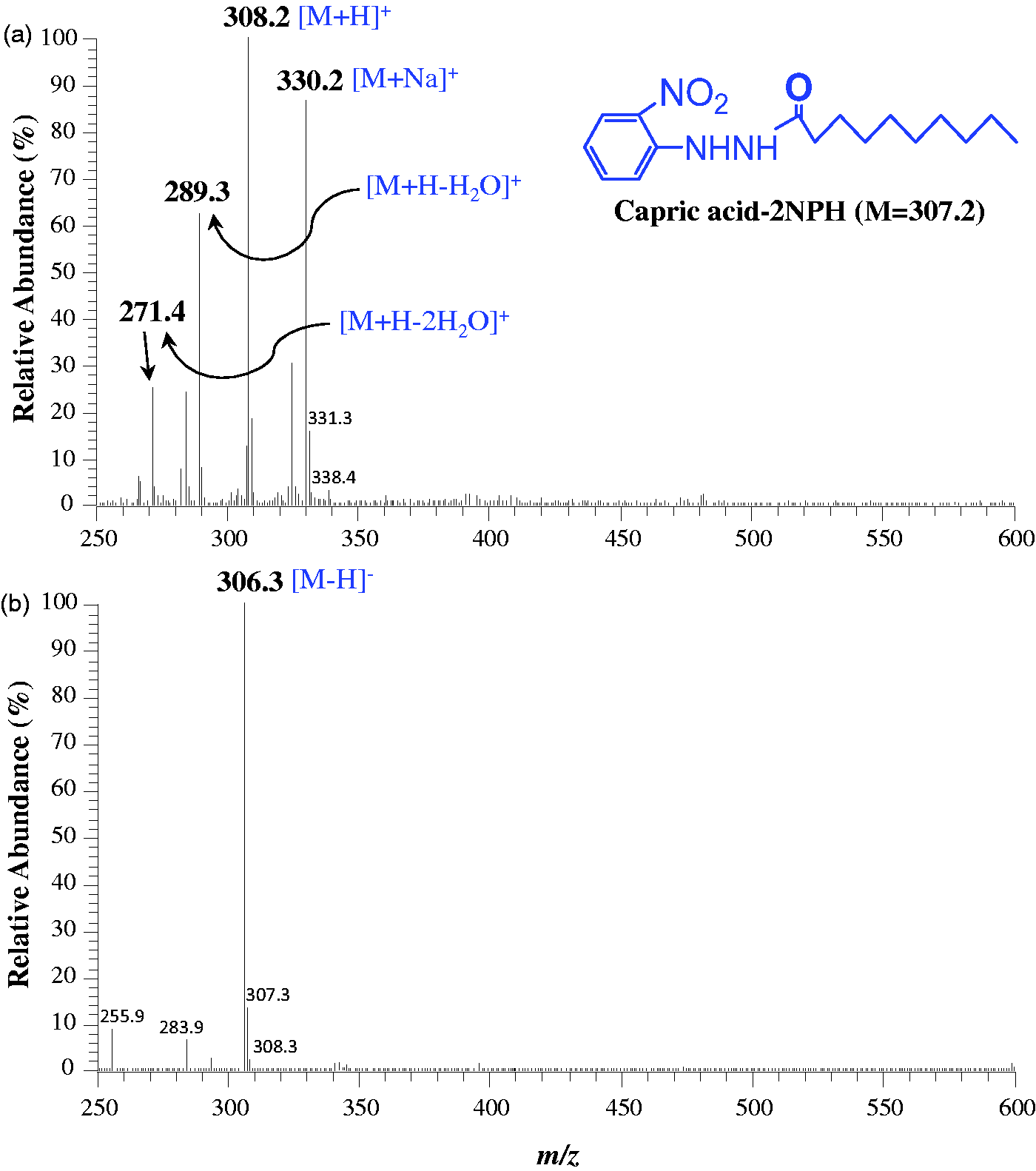
Preparation of standard curve
A stock solution (20 µmol/L) of each FA-NPH was prepared gravimetrically in methanol by measuring its dry weight on an ultrasensitive electro-balance (Cubis® ultramicro balance, Sartorius, Göttingen, Germany). Working standards for HPLC analysis were prepared by mixing standard solutions of FA10:0-NPH, FA14:0-NPH, FA16:0-NPH, and FA18:0-NPH to give the final concentration of each 0.1, 1.0, 2.0, 4.0, 6.0, 8.0, 20 µmol/L. Each standard solution also contained 1 and 4 µmol/L of IS FA11:0-NPH and FA17:0-NPH, respectively. The standard solutions were stored at –20℃ and allowed to equilibrate at room temperature before use. Ten microliters of each standard solution was injected into HPLC. The calibration curves were constructed by plotting the peak area ratio of each FA-NPH standard to the IS (FA11:0-NPH for FA10:0-NPH and FA14:0-NPH, and FA17:0-NPH for FA16:0-NPH and FA18:0-NPH) against its concentration.
Specimen
Fasting blood sample (3 mL) was collected from five healthy human volunteers (three male and two female; mean age ± SD, 31 ± 9.3 years). In another 106 healthy Japanese volunteers (44 male and 62 female; mean age ± SD, 21.9 ± 3.2 years), non-fasting blood samples (3 mL) were collected irrespective of dietary intake. Written informed consent was obtained from all study subjects. Plasma was separated from all of the samples within 30 min of collection and stored at –80℃ until used. Ethics review boards of the Faculty of Health Sciences, Hokkaido University and of Oita University, Faculty of Medicine approved this study protocol.
Assay of FA in plasma
For the derivatization of FA with 2-NPH, a method established by Miwa et al. 11 was used with the following modifications. For the FA10:0 assay, 50 µL of plasma was mixed with 10 µL of FA11:0 (IS, 2 nmol), while for FA16:0 and FA18:0 assay, 10 µL of plasma was mixed with 25 µL of FA17:0 (IS, 5 nmol). A total of 100 µL of KOH (15%, w/v in ethanol:water 50:50) was then added and the mixture was heated at 80℃ for 20 min to ensure complete saponification of triglycerides present in the samples. This was followed by the addition of 200 µL of each 2-NPH·HCL (20 mM in ethanol) and 1-EDC·HCl (0.25 M in ethanol:pyridine 97:3) and heated at 60℃ for 20 min. After the addition of 200 µL of KOH (10%, w/v in methanol:water 50:50), the mixture was further heated at 60℃ for 15 min and cooled. To the resulting mixture, 4 mL of potassium phosphate buffer (pH 4.6) and 3 mL of hexane were added. The mixture was then vortex mixed vigorously for 30 s and centrifuged at 3000 × g for 30 min at 25℃. The upper hexane layer was collected and completely evaporated under vacuum (Tomy centrifugal concentrator, Tokyo, Japan). The residue was dissolved in 200 µL of methanol and filtered using a centrifugal filter device (PVDF 0.1 µm, Merck Millipore Ltd, Carrigtwohill, Ireland). Ten microliters of the extract was injected into HPLC.
Accuracy, precision, recovery, and stability
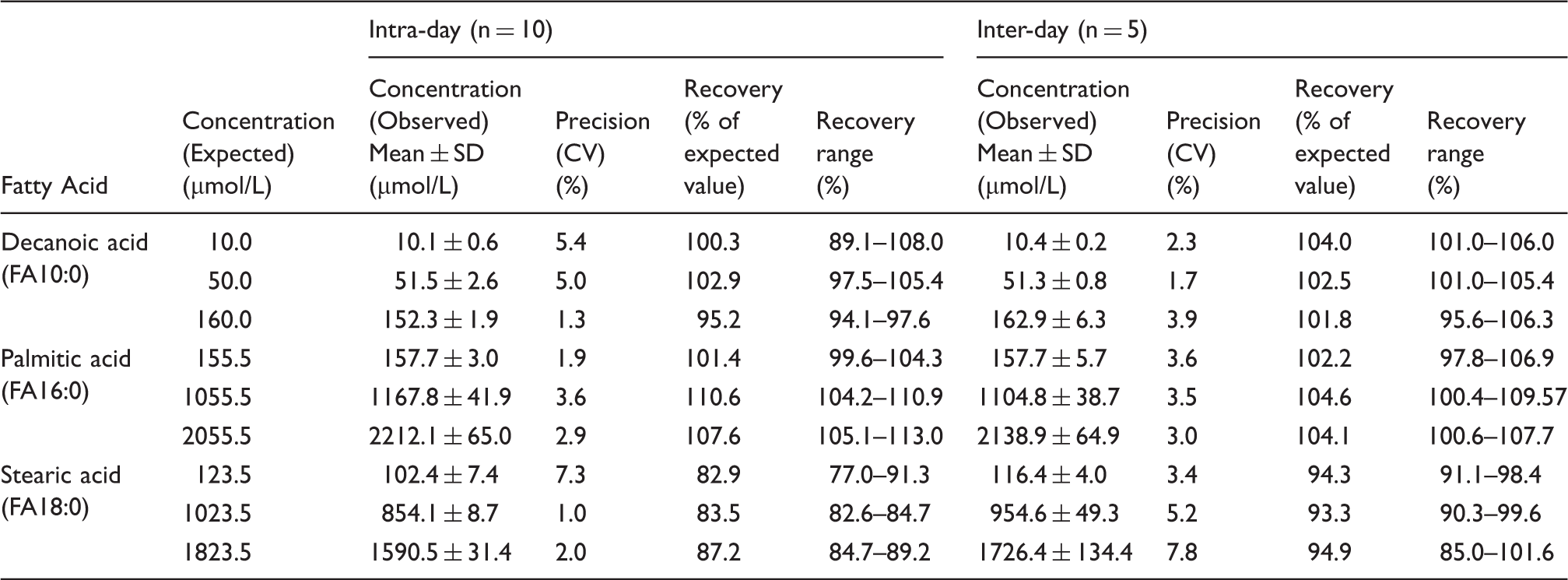
The recovery and reproducibility for the FA assay were investigated by repeating analysis of plasma of healthy human volunteers added with known concentrations of FA10:0, FA16:0, and FA18:0 at three different concentrations—low, normal, and above-normal in the same run and on different days. Since FA10:0 is relatively soluble, pure FA10:0 was directly added in a fasting plasma (10.0 mL) that do not contain FA10:0, to give a final concentration of 200 µmol/L. The mixture was incubated at 37℃ for an hour with mixing several times to ensure complete solubilization. The plasma containing FA10:0 was then diluted to 10, 50, and 160 µmol/L with plasma and 50 µL of each were used for measuring FA10:0 concentration. Additionally, aliquot of these plasma were also stored at –20℃ and analyzed for FA10:0 on every successive month for six months to determine its stability. Solutions of FA16:0 and FA18:0 were added to 10 µL of plasma during the derivatization to give the final concentration of 155.2, 1055.52, and 2055.52 µmol/L of FA16:0 and 123.5, 1023.5, and 1823.5 µmol/L of FA18:0. Intra-assay and inter-assay precision were determined by assaying these spiked human plasma on different days (n = 10) and on the same day (n = 5), respectively.
Liquid chromatography condition
Chromatographic analyses were performed with a Shimadzu Prominence LC-20AD HPLC (Shimadzu Seisakusho, Kyoto, Japan) equipped with a Shimadzu Model CTO-10 A injector and the SPD-M20A photo diode array detector. The absorbance was measured at 400 nm. The separation was performed with C4 Mightysil reversed-phase column (150 mm × 4.6 mm [i.d.]; particle size 5 µm) (Cica Reagent, Kanto chemical Co., Inc, Tokyo, Japan), maintained at 35℃. Gradient elution was performed with water (pH adjusted to 4.0 with 1% [v/v] trifluoroacetic acid) (solvent A) and methanol (solvent B), at a flow rate of 1.0 mL/min. The HPLC gradient conditions were as follows: 0.00–3.00 min isocratic at 60% B, 3.01–30.00 min 60 → 90% B, 30.01–35.00 min isocratic at 100% B to wash away any residual peaks eluting after last peak of interest, and 35.01–40.00 min isocratic at 60% B to re-equilibrate the column.
Results
Separation of fatty acids in plasma by HPLC
The separation of FA-NPH derivatives by reversed-phase HPLC is based on the chemical property of increasing polarity with decreasing chain length of FA. Therefore, by changing the polarity of solvent, the retention time of FA can be adjusted. A series of experiments were performed to identify the optimal HPLC conditions, including types of columns and solvents, and its elution profiles, for the separation of four fatty acids in the plasma (FA10:0, FA14:0, FA16:0, and FA18:0) along with two IS (FA11:0 and FA17:0). The present HPLC method was selected as optimal for the distinct separation of FA without interference with other components in the plasma within elution time of 30 min. For the measurement of FA10:0, 15 min of run time is adequate as both FA10:0 and IS FA11:0 elute within the mentioned time. A typical chromatogram obtained from a mixture of standards and plasma samples was elucidated in Figure 2.
HPLC chromatogram of the 2-nitrophenylhydrazine derivatives of (a) mixture of standard fatty acids, (b) fatty acid from a human plasma spiked with FA10:0 (200 µmol/L), and (c) fatty acids from a fasting human plasma. Peaks: 1, capric (FA10:0); 2, undecanoic (FA11:0); 3, myristic (FA14:0); 4, palmitic (FA16:0); 5, heptadecanoic (FA17:0); and 6, stearic acid (FA18:0).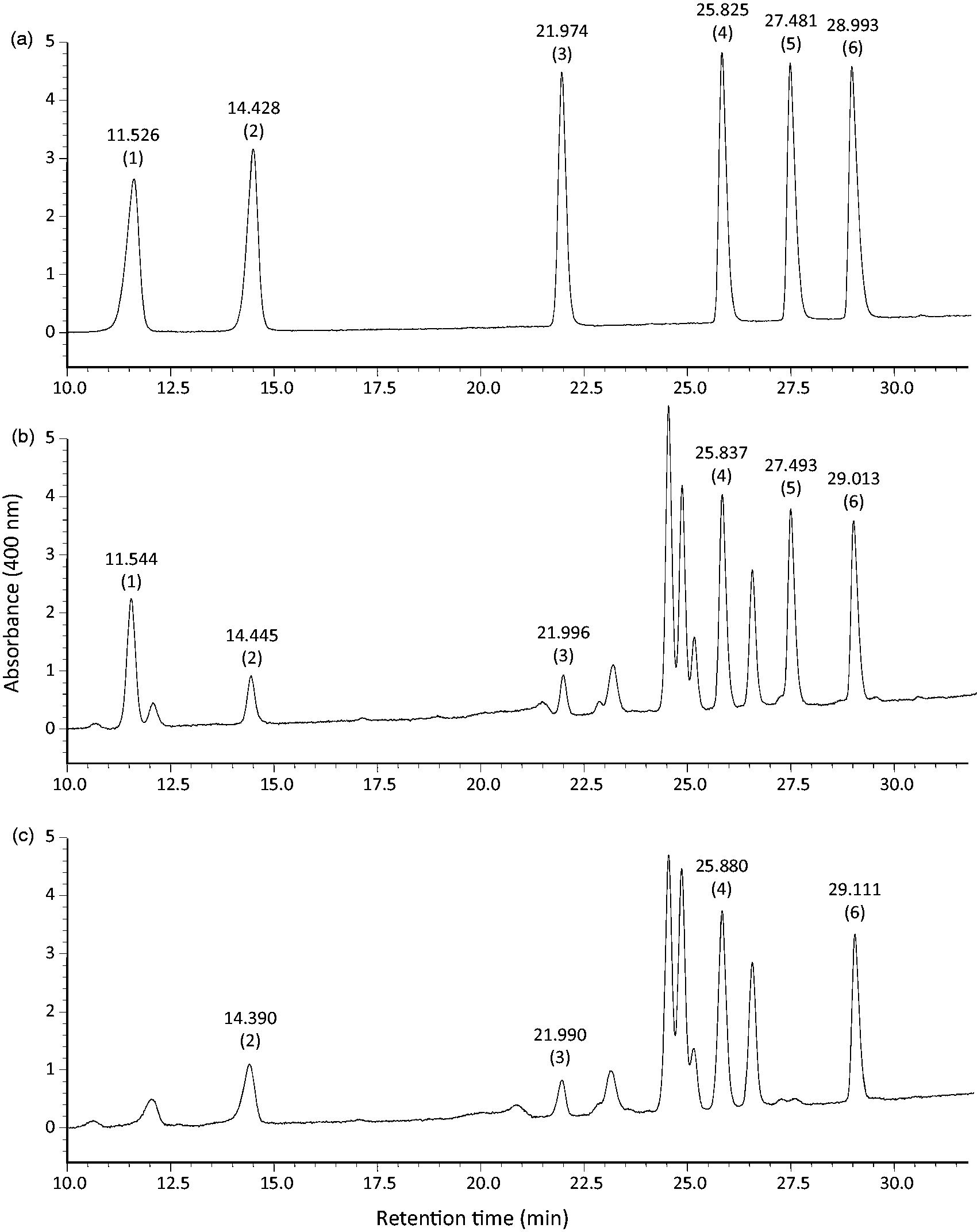
Calibration and sensitivity
An eight-point calibration curve ranging from 1 to 200 pmol per injection for NPH derivatives of FA10:0, FA14:0, FA16:0, and FA18:0 was evaluated. When 10 µL of above-prepared calibration specimen was injected (0.1, 1.0, 2.0, 4.0, 6.0, 8.0, and 20.0 pmol/µL) on the column, the response (y) was linearly related to FA concentration (x). Each calibration curve, as determined by linear regression analysis, showed good linearity with the correlation coefficient (r2) of 0.99829, 0.99800, 0.99988, 0.99862 for FA10:0, FA14:0, FA16:0, and FA18:0, respectively (Figure 3).
Calibration curves for 2-nitrophenylhydrazine derivatives of capric (FA10:0), myristic (FA14:0), palmitic (FA16:0), and stearic acid (FA18:0).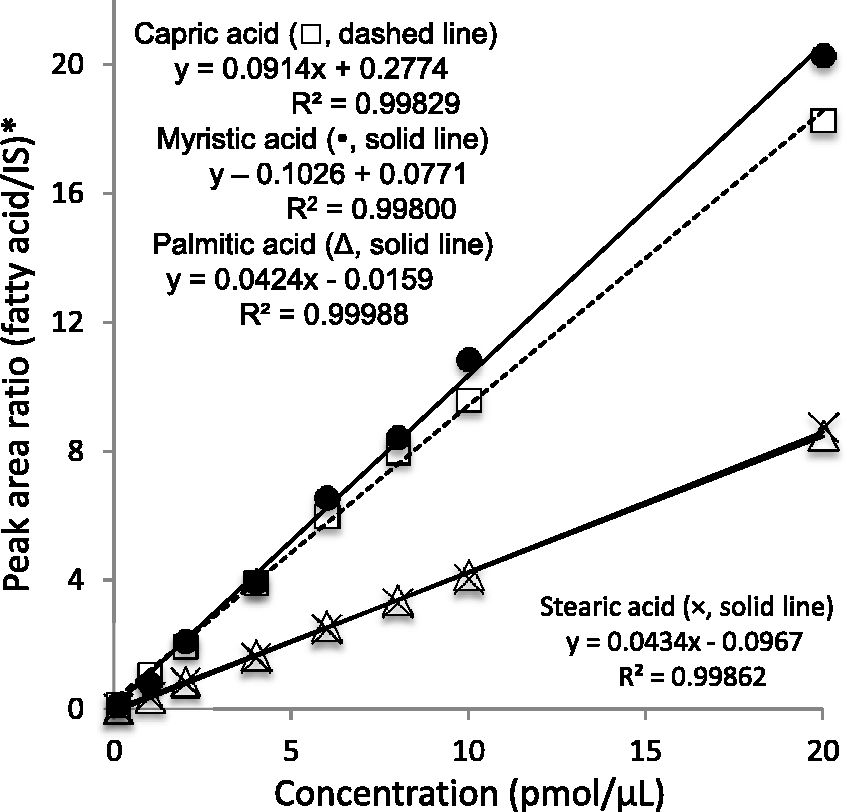
The limits of detection (LOD) were 2 pmol (20 µL of 0.1 pmol/µL) for FA10:0, FA14:0, and FA16:0 at a signal-to-noise ratio (S/N) of 4:1, 4:1, and 3:1, respectively. The LOD of FA18:0 was 1 pmol at S/N 3:1. With our HPLC method, the lower limit of quantification was 3 pmol for each FA at S/N 5:1 per injection to the column.
Recovery and reproducibility
Inter and intra-assay precision and accuracy of three fatty acids in serum evaluated by means of coefficients of variation and recovery of added standards from spiked serum samples (%).
FA10:0 in human plasma
No peak was detected at the elution time for FA10:0 in all fasting plasma of healthy humans (Figure 2(c)), indicating that the normal fasting plasma concentration of this FA is below the detection limit (0.1 µmol/L). The mean plasma concentration of total FA16:0 and FA18:0 in the fasting healthy volunteer (n = 5) were 2092.1 (SD 244.1) and 696.8 µmol/L (SD 76.3), respectively.
Spearman’s correlation between plasma saturated fatty acids concentration (n = 106).
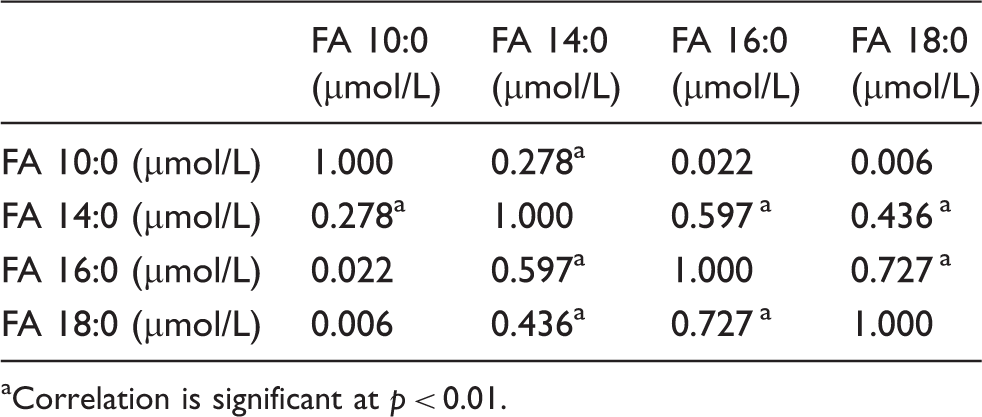
Correlation is significant at p < 0.01.
Discussion
We found our HPLC method for the measurement of FA10:0 to be a simple, accurate, and reproducible method that can be applied to monitor its concentration following MCT therapy. The procedure is relatively non-expensive and rapid, as many samples can be processed in a single batch without undue effort and the reaction products are stable for many days at 4℃. Although the last peak of interest (FA18:0) elutes at about 30 min, it is necessary to run blank for a further 10 min, to provide sufficient time for elution of some unidentified peaks appearing after 30 min. However, for the measurement of FA10:0, 15 min of HPLC run is sufficient. In addition, several other factors contribute to the superiority of our method. Firstly, we used FA11:0 as IS, which has similar physical and chemical properties to FA10:0, in contrast to many reported method using FA17:0, a LCFA as the IS. Secondly, high resolution and isolated peaks for these FA were observed in the chromatogram without interference. Thirdly, the assays have very good linearity from 1 to 200 pmol per injection. Fourthly, both total and free FA10:0 can be determined in the same HPLC condition and derivatization technique with and without saponification, respectively. Lastly, the analytical recoveries were ranging from 95 to 104% with good reproducibility of the assay, making it suitable for the measurement of both free and total FA10:0 for clinical uses. Furthermore, this method can be also used to measure LCFA simultaneously. However, the recovery of FA18:0 was found to be lower than those of FA10:0 and FA16:0 (Table 1). The relatively low recovery of FA18:0 was also reported by Miwa. 12 The low recoveries of FA18:0 might be attributed to a possibly less reactivity of FA18:0 with 2-NPH than those for shorter chain fatty acids. When the labeling with 2-NPH is used for the measurement of short- to LCFA, appropriate IS, according to the length of FA might be necessary.
FA10:0 was undetectable in the fasting plasma of healthy volunteers as consistent with other studies.15,16 However, concentrations of FA16:0 and 18:0 in these subjects were within the normal and acceptable ranges, which further add to the reliability of this method. FA10:0 concentration in the non-fasting samples were negligible. Only 27 (25.5%) of human volunteers have FA10:0 concentration above 0.5 µmol/L, with the maximum of 1.6 µmol/L, which is still more than 1000 times lower than the concentration of FA16:0, most predominant FA in human plasma. There is scanty of data available about plasma concentration of FA10:0. It is believed that majority of dietary MCFA are carried to liver through portal circulation and are completely metabolized within it. Therefore, systemic availability of FA10:0 is negligible unless MCT is used therapeutically. Existence of FA10:0 in trace amount in some non-fasting individuals in this study may reflect dietary FA10:0 that escape hepatic utilization. The most possible source of FA10:0 in Japanese diet is milk and its products. Therefore, it appears that plasma concentration of FA10:0 depends on dietary status. Communities where coconuts and its oils are extensively taken orally, such as south India, particularly Kerala (Literally “Land of coconut palms”), possibly have higher plasma concentrations of FA10:0.
GC approach for measuring FA10:0 specifically have been reported.15,17 Haidukewych et al. reported that the concentrations of FA10:0 are below detection limit in plasma. 15 In contrast, Onkenhout et al. previously reported relatively higher concentration of plasma FA10:0 in control children using GC. 18 The higher concentration may be linked with higher consumption of milk in children compared to adults. HPLC-based assay for the measurement of FA10:0 has been described by Dean et al. 19 They used α-p-dibromoacetophenone for the derivatization of MCFA and determined FA10:0 in plasma after treating children with MCT diet but not in healthy subjects. The derivatization of FA with NPH followed by UV detection as described by Miwa et al. has been implicated for the determination of short-chain FA.20,21 However, use of this labeling technique for the specific measurement of FA10:0 in plasma has not been reported yet. Our method can be particularly valuable to measure plasma FA10:0 during MCT therapy in order to acquire precise information about the therapeutic target of MCFA. As being readily digested by acid stable gastric lipase and predominantly absorbed via portal vein, MCT have been historically used in the treatment of pancreatic insufficiency and fat malabsorption syndrome. In the recent years, the interest of dietary intervention with MCT is increasing to benefit varieties of clinical conditions including obesity,3,22,23 metabolic syndrome, 6 insulin resistance,24,25 liver diseases, 26 and cardiac diseases. 5 Furthermore, studies are reveling that MCFA incorporates into the cellular lipids at a lower rate than LCFA and induce a lower accumulation of triglycerides. 27 This phenomenon is particularly significant in neutral lipids storage disorders. In addition, it appears that the mobilization of cytoplasmic MCT is independent of the action of adipose tissue triglyceride lipase (ATGL). 27 Therefore, it can be speculated that MCT therapy reduces the lipids accumulation in ATGL mutated cells. It is of significance to monitor plasma free and total FA10:0 concentration, and its association with clinical signs and symptoms in patients receiving dietary therapy with MCT, which would provide scientific evidence about mechanism explaining the benefit of dietary therapy.
In conclusion, our studies describe an improved method for the separation, derivatization, and quantification of FA10:0 in plasma using HPLC with sufficient accuracy and reproducibility. FA10:0 can be detected in trace amount in healthy human during non-fasting state. Application of this method may be valuable in the monitoring of FA10:0 concentration during therapeutic use of MCT.
Footnotes
Declaration of conflicting interests
None declared.
Funding
This study was financially supported by research grants for rare and intractable disease from the Ministry of Health, Labor, and Welfare of Japan, partially supported by the Center of Innovation Program from Japan Science and Technology Agency, JST, by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, Japan, partially by the Regional Innovation Strategy Support Program, Sapporo Health Innovation “Smart-H,” of the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Ethical approval
Ethics review boards of the Faculty of Health Sciences, Hokkaido University (Ref. 14-17) and of Oita University Faculty of Medicine (Ref. 715) approved this study protocol.
Guarantor
HC.
Contributorship
RS, SPH, and HC researched literature and conceived the study. SPH, AS, SY, KH, HC, and RS were involved in study design and discussion. RS, SPH, HI, SH, ST, HF, ST, and NU were involved in sample collection, sample preparation, synthetic standards, NMR, mass spectrometric, HPLC, and data analysis. RS wrote the first draft of the manuscript. All authors reviewed the manuscript and approved the final version.