
Abstract
There is growing interest in the role of fibroblast growth factor 23 (FGF23) in various diseases of disordered mineral metabolism. In chronic kidney disease (CKD), where biochemical evidence of mineral disturbances is especially common, FGF23 measurement has been advocated as an early and sensitive marker for CKD-related bone disease. In this setting, FGF23 analysis may also improve the discrimination of risk of adverse renal and cardiovascular outcomes and aid targeting of those patients that are likely to benefit from interventions. Nonetheless, while the physiological relevance of FGF23 in the control of mineral metabolism is now firmly established, relatively little attention has been paid to important preanalytical and analytical aspects of FGF23 measurement that may impact on its clinical utility. Here we review these issues and discuss the suitability of FGF23 testing strategies for routine clinical practice. The current ‘state-of-the-art’ enzyme-linked immunosorbent assay methods for FGF23 measurement show poor agreement due to differences in FGF23 fragment detection, antibody specificity and calibration. Such analytical variability does not permit direct comparison of FGF23 measurements made with different assays and is likely to at least in part account for some of the inconsistencies noted between observational studies. From a clinical perspective, the lack of concordance has implications for the development of standardized reference intervals and clinical decision limits. Finally, the inherent assay-dependent biological variability of plasma FGF23 concentration can further complicate the interpretation of results and the design of FGF23-based testing protocols. Currently, it would be premature to consider incorporating FGF23 measurements into standard testing repertoires.
Keywords
Background
Fibroblast growth factor 23 (FGF23) is a key bone-derived regulator of phosphate and vitamin D metabolism. In the proximal renal tubule, FGF23 acts to (1) promote phosphaturia by down-regulating the expression of luminal sodium-dependent phosphate transporters and (2) reduce synthesis of 1,25-dihydroxyvitamin D by down-regulating 1α-hydroxylase and up-regulating 24-hydroxylase activity (Figure 1).
1
The classical endocrine actions of FGF23 are mediated by activation of binary receptor complexes formed from one of several FGF-receptor (FGFR) isoforms and α-klotho.
2
α-Klotho enhances the binding affinity of FGF23 for FGFR and confers tissue specificity to FGF23 action due to its limited expression (predominantly the kidneys and parathyroid glands). Although the control of FGF23 production remains poorly understood, a complex interplay between systemic and local factors would appear to allow the coordination of bone buffering capacity with renal phosphate handling.
Engagement of the FGFR-α-klotho receptor complex in the proximal tubular cells leads to receptor autophosphorylation and sequential phosphorylation of ERK1/2, SGK1 and scaffolding protein NHERF1. Activation of NHERF1 results in luminal sodium-dependent phosphate transporter NPT2a internalization and lysosomal degradation and hence, reduced phosphate reabsorption. FGF23 signalling in distal tubular cells may lead to the elaboration of a paracrine factor ‘signal’ that likewise stimulates down-regulation of luminal sodium-dependent phosphate transporters. FGF23 also down-regulates expression of 1α-hydroxylase and up-regulates expression of 24-hydroxylase thereby lowering 1,25(OH)2D3 concentrations. Dashed lines indicate putative pathways/mechanisms.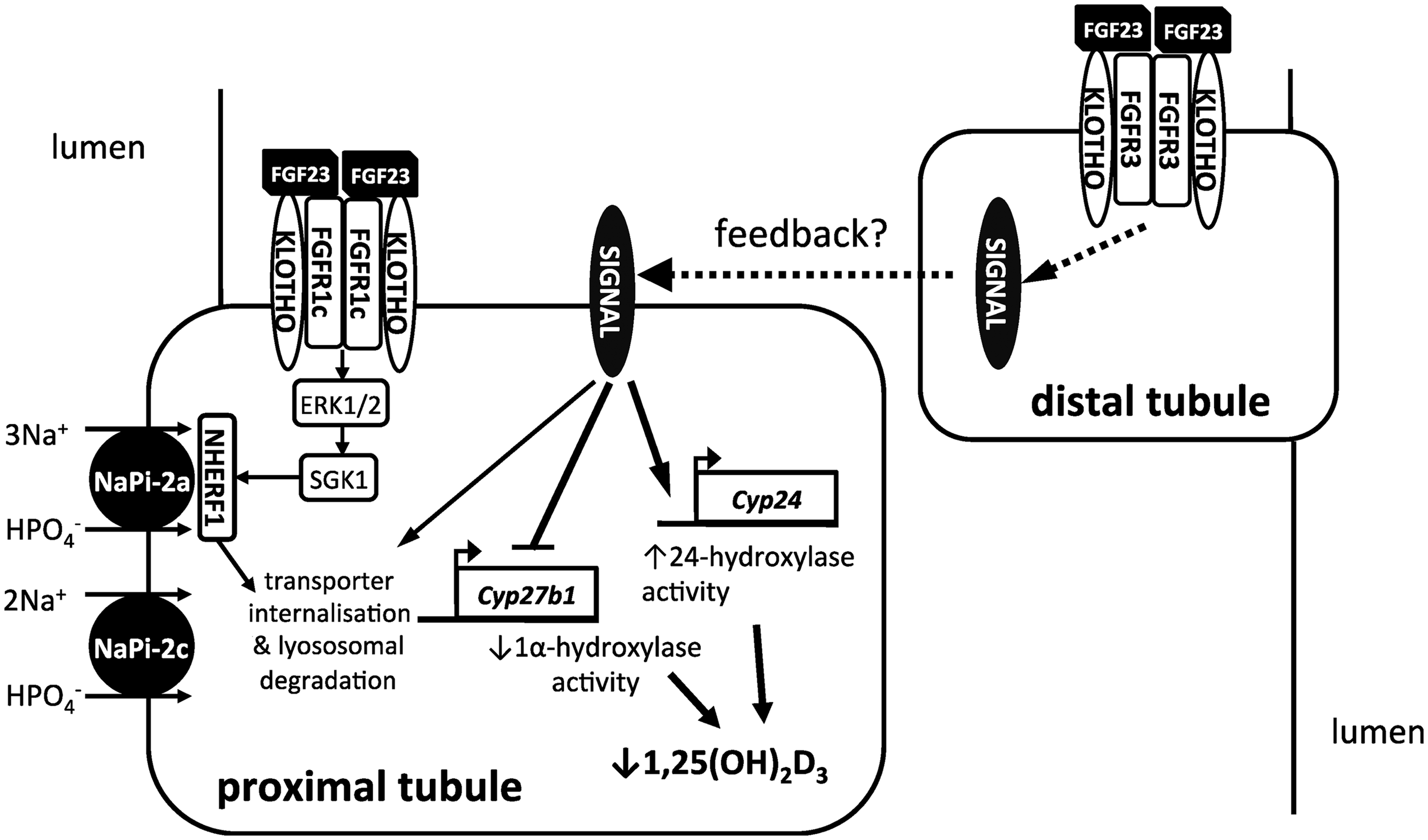
Abnormal plasma FGF23 concentrations were first described in patients with rare renal phosphate-wasting syndromes such as autosomal dominant hypophosphataemic rickets (ADHR) and tumour-induced osteomalacia (TIO),3,4 but have subsequently been observed in a number of hereditary and acquired hyper- and hypophosphataemic disorders. Over the last 5 years FGF23 has been subject to the most intense study in the arena of chronic kidney disease (CKD), where it has transformed the understanding of mineral and bone-related pathophysiology.
FGF23 in CKD
Classically, the progressive loss of functioning nephrons and resultant retention of phosphate has been viewed as the key driver of secondary hyperparathyroidism and the constellation of skeletal and mineral derangements in this setting – now defined by Kidney Disease: Improving Global Outcomes as the chronic kidney disease-mineral bone disease (CKD-MBD) disorder. 5 Recent evidence, however, has challenged this paradigm and the assumptions on which it is originally based. In CKD, plasma FGF23 concentrations increase early, and quite markedly, with deteriorating renal function, probably before phosphate and parathyroid hormone (PTH). 6 It is unclear whether this represents an adaptive response to phosphate retention, changes in bone mineralization and end-organ resistance due to α-klotho down-regulation or is directly and pathomechanistically related to CKD progression. Bone biopsies of patients with very mild renal impairment (CKD Stage 2), and without overt mineral handling problems already show increased FGF23 expression in osteocytes. 7 Interestingly, patients with polycystic kidney disease, but a relatively preserved glomerular filtration rate (GFR), also have increased plasma FGF23 concentrations. 8 While after unilateral nephrectomy, otherwise healthy kidney donors show small but significant increases in circulating FGF23 and increased urinary fractional phosphate excretion. 9 This has led to the suggestion that kidney damage per se might elaborate a signal to bone stimulating FGF23 secretion. 10 Consistent with this hypothesis, epidemiological data show that plasma phosphate concentrations may actually fall in the initial stages of disease (estimated GFR 60–69 mL/min/1.73 m2), at a time when plasma FGF23 concentration and fractional excretion of phosphate are increased.8,10–13 Indeed, very recent data imply that some patients with CKD may actually be in negative phosphate balance due to dietary insufficiency, reduced gastrointestinal absorption and potentially, an inappropriate degree of phosphaturia. 14 Sustained increases in FGF23 secretion also suppress renal 1,25-dihydroxyvitamin D synthesis and contribute to the development of secondary hyperparathyroidism. 15 In patients with end-stage renal disease (ESRD) undergoing dialysis therapy, plasma FGF23 concentrations often reach many orders of magnitude higher than those found in healthy adults. 16
Numerous epidemiological studies have reported a robust association between higher plasma FGF23 concentrations and poor patient outcome in CKD populations, independent of traditional cardiovascular and renal risk factors. In addition to predicting CKD progression,17–21 elevated plasma FGF23 concentrations have been independently associated with increased cardiovascular event rates and mortality risk in dialysis patients,16,18,22 predialysis CKD cohorts18,19,23,24 and in patients with coronary artery disease without significant renal impairment. 25 Higher plasma FGF23 concentrations have also been associated with increased risk of allograft loss after kidney transplant. 23 Risk reclassification analyses suggest that FGF23 ascertainment may add value to risk estimates based on established clinical parameters.21,26 Landmark work by Faul and colleagues has provided compelling evidence that very high concentrations of FGF23 may have direct ‘off-target’, α-klotho-independent, effects on the heart, inducing pro-hypertrophic gene programs, resulting in left ventricular hypertrophy (LVH) in mice. 27 Chronic exposure of the cardiovascular system to sustained high concentrations of FGF23, as seen in CKD and other states of FGF23 excess, may therefore have direct pathogenic sequelae. Thus, FGF23 has been elevated from the status of mere risk marker, to a potentially causative and modifiable risk factor. On the other hand, there is some evidence that activation of FGFR/α-klotho pathways might be protective to the vasculature, inhibiting the osteogenic transformation of vascular smooth muscle cells and reducing arterial matrix calcification.28–31 However, these protective effects upon vessels remain controversial, since vascular calcification is not a prominent feature of transgenic animals over-expressing FGF23 and recent data would appear to suggest that FGF23 has no independent effect on phosphate-driven vascular calcification.32,33 Moreover, unlike the consistent relationship between higher circulating FGF23 concentrations and LVH, the association with vascular calcification seems variable and phosphate dependent.32,34–36 FGF23 has also recently been shown to suppress expression of renal angiotensin-converting enzyme 2;37,38 however, its effects on the renin–angiotensin system, which may also present a credible mechanistic link between renal and cardiovascular disease pathways, remain unexplored.
Potential therapies targeting FGF23 excess
Presently, there are several experimental strategies for reducing excess FGF23 bioactivity, principally through dietary phosphate restriction or use of oral phosphate binders, or by antagonism of FGFR signalling or direct immunochemical neutralization of FGF23 protein. Although inhibition of FGFR signalling might be a promising strategy in FGF23-mediated hypophosphataemic bone disorders (e.g. NVP-BGJ398, a novel selective, pan-specific FGFR inhibitor), 39 there are currently no data to suggest that any of the aforementioned approaches yield any clinical benefits to patients with renal disease. On the contrary, in the setting of predialysis CKD, FGF23 neutralization or phosphate-lowering strategies may prove counterproductive, disturbing the delicate adaptive response to changes in mineral handling. Indeed, although FGF23 neutralization has been demonstrated to limit the progression of secondary hyperparathyroidism and normalize bone turnover in uraemic rats, it did so at the expense of hyperphosphataemia, increased aortic mineralization and excess mortality. 40 In patients with CKD, the clinical efficacy of phosphate-lowering strategies with oral binders also remains unproven, and available data are inconsistent with respect to their effects on circulating FGF23 concentrations. Definitive findings from larger, better-powered clinical trials such are needed to clarify this situation.
Potential utility of FGF23 testing
In current clinical practice, FGF23 testing may occasionally be requested in the investigation of patients with rare chronic renal phosphate-wasting syndromes, but availability is limited to specialist centres. More widespread measurement of FGF23 has been advocated in the setting of CKD. Here, FGF23 is regarded as an early and sensitive marker of disordered mineral metabolism, which may facilitate more effective discrimination of the risk of related adverse outcomes and targeting of those likely to benefit from intervention.6,41 Crucially, with respect to mineral disorders, it has been suggested that FGF23 measurement may be superior to existing contemporaneous measures of mineral metabolism such as PTH and phosphate.6,41 Beyond the CKD setting, FGF23 testing has been advocated in the evaluation and management of patients with disturbances in plasma phosphate concentrations.42,43 FGF23 measurement during selective venous sampling may also be helpful as an adjunct to functional imaging for the localization of tumours in patients with TIO. 44 Such applications should, however, be considered experimental at this stage, as their clinical value has not been conclusively demonstrated.
Despite growing interest in the use of FGF23 testing in routine clinical practice, remarkably little consideration has been given to important preanalytical and analytical aspects of FGF23 measurement. Studies concerning syndromes of FGF23 excess and deficiency, basic hormone function and regulation, and epidemiological observations have been reviewed elsewhere and the reader is directed to these articles for a more detailed discussion of these topics.45–47 This article will briefly review relevant FGF23 biochemistry and then focus on accumulating data that shed some light on the interpretation and utility of FGF23 measurements.
FGF23 – structure and regulatory pathways
The FGF23 gene has three exons and spans ∼10 kb of genomic DNA on human chromosome 12p3.3.
48
Physiologically, FGF23 is highly expressed in osteocytes and osteoblasts,
49
but is also sparingly expressed in heart, liver, skeletal muscle, mammary glands and brain.
48
The significance of extra-skeletal FGF23 production, if any, is unknown. Although incompletely understood, FGF23 synthesis in bone is controlled by the interplay between a number of systemic and local factors, which regulate expression at the level of transcription and by modification of protein stability (Figure 2). Major positive regulators of FGF23 expression include 1,25 dihydroxyvitamin D, phosphate, PTH and the proteolytic cleavage product of membrane-bound α-klotho (reviewed in Martin et al.46). 1,25 dihydroxyvitamin D-mediated regulation also appears to be modulated by a number of secondary factors including leptin and interleukin-6.
50
Iron deficiency stimulates FGF23 expression, potentially via hypoxia inducible factor-1α pathways.
51
Calcium deficiency, on the other hand, appears to suppress FGF23 secretion,
52
again by unknown mechanisms. Local bone-derived factors such as dentin matrix protein-1 (DMP-1) and phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) have a suppressive effect on FGF23 transcription,
53
while phosphate, iron status and DMP-1 also appear to modulate FGF23 bioactivity via differential regulation of the degradation pathway.
Regulation of FGF23 secretion from bone cells. The synthesis and degradation of FGF23 in osteocytes and osteoblasts is regulated by various systemic and local factors (left-hand side of panel). FGF23 is synthesized as a pro-protein. After cleavage of the 24 N-terminal signal peptide, a fraction of FGF23 undergoes O-linked glycosylation by GALNT3 (UDP-N-acetyl-α-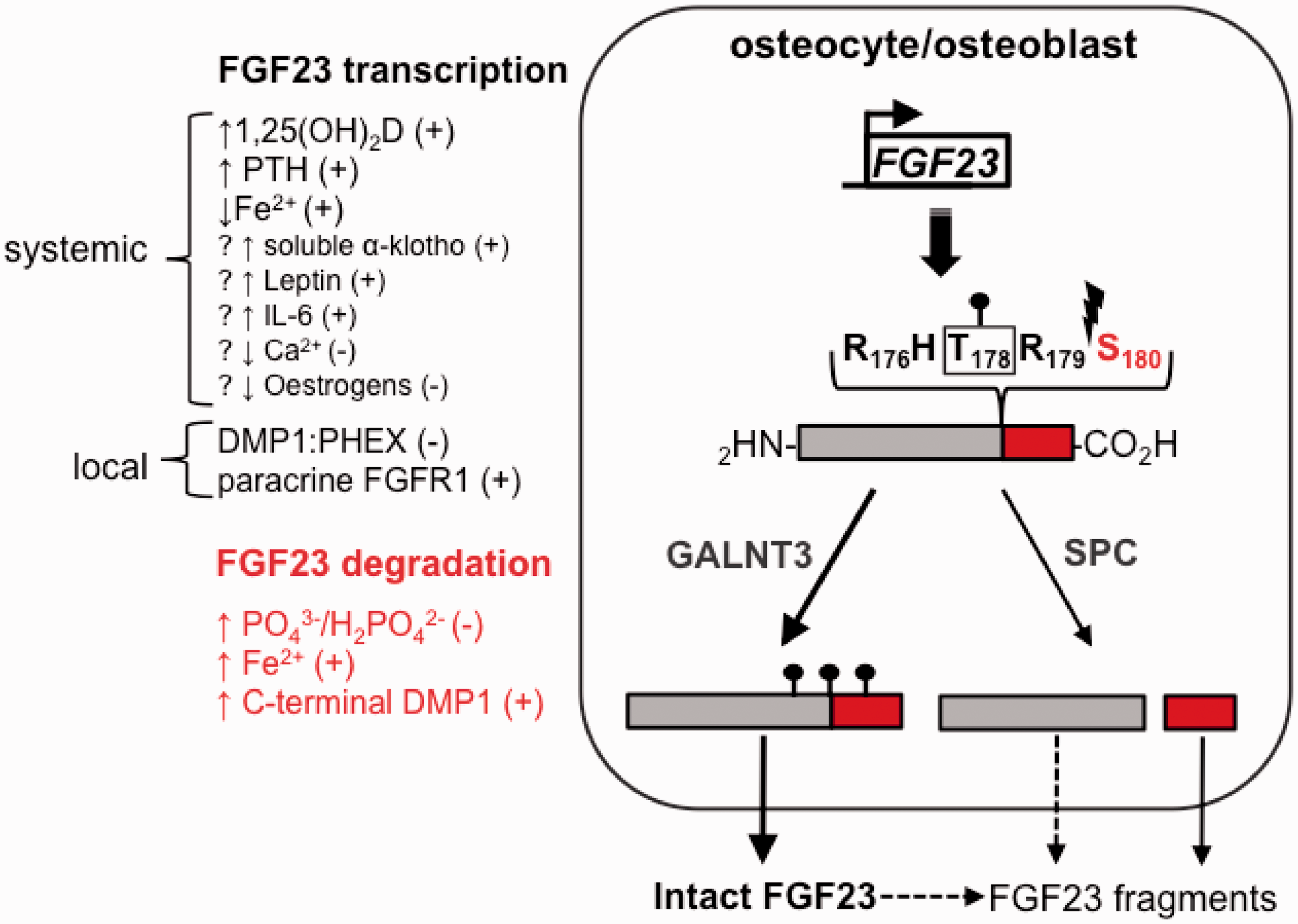
FGF23 is synthesized as a 251-amino acid precursor, comprising a 24-residue signal peptide, an N-terminal FGF-like homology domain (25–179) and a unique C-terminal sequence (180–251) that mediates high-affinity binding to the FGFR:α-klotho receptor complex.
54
After removal of the signal peptide, FGF23 undergoes modification by disulphide bond formation (C95–C113) and O-glycosylation by uridine diphosphate-N-acetyl-alpha-
Cleavage at the PC consensus site abolishes the phosphaturic action of intact FGF23 on the kidney. This is primarily due to loss of the receptor binding motif located within the C-terminal portion of the molecule, 57 but, importantly, is also because the C-terminal fragments generated by this processing may compete with intact protein for target cell receptor binding, thus antagonizing the action of intact FGF23 and its downstream biological effects. 58 Proteolysis is therefore considered to be a critical level of regulation of FGF23 bioactivity. Furin, PC5 and PC2, in consort with its essential binding cofactor, 7B2, have been implicated as potential mediators of FGF23 cleavage in bone.56,59 Furin and PC5 have a wide tissue expression, with activity predominantly localized to the trans-Golgi network, cell surface and extracellular matrix, and are responsible for the processing of various growth factors, adhesion molecules and receptors. 60 Apart from bone, PC2 expression is mainly confined to neuroendocrine cells and is localized to the immature and dense-core granules within the acidic regulated secretory pathway.
Missense mutations in FGF23 at R176 and R179, as found in patients with ADHR, give rise to a mutant protein that is resistant to cleavage and undergoes very limited processing by PC. 61 Such gain-of-function mutations augment the circulating concentration and half-life of intact protein, increasing its biological potency, manifesting as a renal phosphate-wasting syndrome in affected individuals. 61 ADHR has a variable penetrance, and FGF23 concentrations are usually only elevated in patients with active disease. 62 The variable onset of ADHR appears to relate to the requirement for coexistent iron deficiency, which augments FGF23 expression by the osteocyte. 51 The effect of increased FGF23 synthesis due to iron deficiency, in combination with restricted PC-mediated inactivation, results in increased biologically active FGF23 and the classical hypophosphataemic rickets phenotype.
Co-precipitation experiments suggest that at least a fraction of the circulating FGF23 may be bound to soluble α-klotho. 63 Soluble α-klotho derives from two principal sources; endoproteolytic cleavage and ectodomain shedding of membrane bound KL (cKL), and a secreted form produced by alternative splicing on the KL gene (sKL). 64 The cKL form appears to be the predominant species in human circulation and a paracrine mediator of renal phosphate handling in animal models. 65 cKL may serve as a ‘portable’ co-receptor allowing FGF23-induced signalling in cells that do not ordinarily express the membrane-bound form. 66
Although the balance between synthetic and degradation pathways would appear to govern FGF23 bioactivity, there is a paucity of data pertaining to its metabolism and clearance: it is not known how or where FGF23 or its fragments are cleared from the circulation. Using Western blotting, Larsson et al. observed a small amount of intact FGF23 and fragments in the urine of a healthy individual and a single haemodialysis patient with residual renal function (although not corresponding to fragments generated by cleavage at the PC consensus site), but concluded that contribution of urinary excretion to total body FGF23 clearance was likely to be negligible. 67 Similarly, in patients with ESRD undergoing peritoneal dialysis, dialytic clearance of FGF23 has been reported to contribute minimally to elimination. 41 During a standard 4-h haemodialysis session, serum intact FGF23 concentrations have been reported to fall by up to 26%, but this reduction appears highly variable and rebounds quickly before the next session. 68 It remains to be determined whether renal tubular uptake and/or metabolism of FGF23 is an important pathway of FGF23 degradation and whether this may be modified in the context of renal dysfunction. It is, however, also important to recognize that these clearance studies are based solely on the immunochemical detection of FGF23 species and may not account for non-immunoreactive FGF23 protein.
The elimination of intact FGF23 following excision of tumours in patients with biopsy-proven TIO suggests a half-life in plasma of between 22 and 94 min. 69 Once again, however, it is not clear whether tumour-derived FGF23 is structurally or immunologically similar to that synthesized physiologically by osteocytes. Indeed, ectopically expressed proteins often differ in their glycosylation pattern, a factor shown to be important in FGF23 stability. Interestingly, analysis of data from the combined intravenous infusion of fluorescently labelled recombinant intact and C-terminal FGF23 into Sprague–Dawley rats suggests that full-length intact protein is cleared from the circulation almost twice as fast as the C-terminal fragment, with plasma half-lives of 6 and 12 min, respectively. 70 While recombinant mutant FGF23, harbouring ADHR-related mutations R176Q and R179Q, displays a plasma half-life approaching 3 times that of the wild-type protein (∼17 min). Notably, these experiments also provided evidence of rapid (<5 min) proteolysis of exogenous intact FGF23 together with de novo generation of C-terminal fragments within the circulation. These studies complement ex vivo evidence showing rapid degradation of intact FGF23 in whole blood within 2 h of venepuncture, a process that is abrogated by the addition of broad-spectrum protease inhibitors. 71
FGF23 measurement
Currently, there are two formats of immunometric enzyme-linked immunosorbent assay (ELISA) available commercially for measurement of human FGF23 in plasma or serum: ‘intact’ assays (iFGF23) recognizing distant epitopes flanking the PC cleavage site, 72 and the ‘C-terminal’ assay (cFGF23), which recognizes two distinct epitopes both with the C-terminal portion of the molecule, which therefore detects both intact FGF23 and C-terminal fragments. 73
The cFGF23 assay from Immutopics Inc. (San Clemente, CA, USA), now in its second generation, uses two polyclonal goat antibodies that bind epitopes mapped within residues 186–206 and 225–244. The Immutopics iFGF23 assay uses the same C-terminal specific capture antibody as used in the cFGF23 assay and a horseradish peroxidase-conjugated N-terminal specific detection antibody recognizing residues 51–69. The iFGF23 assay from Kainos Laboratories Inc. (Tokyo, Japan) utilizes two murine monoclonal antihuman FGF23 antibodies, FN1 (recognizing N-terminal fragments) and FC1 (recognizing C-terminal fragments) as capture and detection antibodies, respectively. Epitope mapping data for these antibodies are not available. Both assays from Immutopics use a single-step incubation of sample with capture and detection antisera, whereas sample is reacted with capture and detection antibodies sequentially in the standard two-step assay from Kainos.
All three assays are calibrated using recombinant human FGF23 expressed in murine myeloma cell lines. Units of measurement differ, however, with the iFGF23 assays being calibrated in units of mass concentration (picograms per millilitre), whereas the cFGF23 has a readout in relative units (RU) per millilitre. There is currently no commercial source of purified, native human FGF23 or international reference preparation (IRP) of synthetic FGF23. While the Kainos and Immutopics kits have been widely used and cited in the literature, there is very little information pertaining to the iFGF23 assay recently marketed by Millipore (Billerica, MA, USA). The Millipore assay purports to have a wider functional analytical range; however, this comes at the expense of poor sensitivity at low concentrations. 74 Of note, the limited functional analytical range of the available commercial assays is a particular problem for states of FGF23 excess (e.g. dialysis patients where concentrations can exceed 100,000 RU/mL), as this necessitates very large dilutions to bring concentrations within the working range. Furthermore, the appropriate dilution factor must be determined empirically, which can lead to excessive repeat testing. Shimizu et al. have recently developed an automated chemiluminescent iFGF23 assay using the monoclonal antibodies employed in Kainos assay. 75 Such methodology has clear advantages over the ELISA-based kits that have been used to date; needing smaller samples volumes (10 µL vs. 100 µL), wider analytical range (1–15,000 pg/mL vs. 10–800 pg/mL) and shorter run times (20 min vs. 3 h).
Formally defined reference intervals for plasma FGF23 concentrations have been described in adult and paediatric populations using Immutopics kits, but not for the Kainos or Millipore iFGF23 assays.76,77 In healthy adults, the 95% reference limits for plasma iFGF23 is 11.7–48.6 pg/mL and for cFGF23 21.6–91.0 RU/mL. 77 During infancy and adolescence, cFGF23 concentrations are significantly and inversely correlated with age and median concentrations only approach adult concentrations at 4 years of age. 76 It should be noted, however, that none of the aforementioned assays have been validated for clinical use and are marked for research use only.
Evaluating the potential clinical utility of routine FGF23 testing
Although plasma FGF23 measurement is already occasionally performed in the assessment of patients with unexplained hypophosphataemia, more general application in clinical practice necessitates further consideration. In the evaluation of any new biomarker, a number of preanalytical, analytical and clinical issues need to be thoroughly examined. With respect to the preanalytical phase, an ideal biomarker would be stable, undergo little degradation and show minimal variability diurnally and longitudinally. In terms of analysis, there must be accessible high-throughput methodology that is accurate, reproducible and affordable. Finally, the information gleaned by measurement should add to, or improve upon existing tests, aid risk assessment or enhance patient management. Promisingly, it has been suggested that FGF23 and methods for its determination show many of these important attributes.6,41,78–82 It cannot be denied that FGF23 measurement shows great promise as an indicator of poor outcomes in patients with CKD; few biomarkers in recent memory have been shown to be so robustly associated with hard endpoints with minimal confounding. However, the proposed use of FGF23 testing to target therapy in patients with CKD, a disorder with a prevalence of 8–16%, carries with it significant economic and practical implications for the clinical laboratory and health services alike. The following discussion appraises some of the important issues surrounding FGF23 measurement and assesses the implications for testing in research and clinical practice.
Preanalytical issues – stability and biological variability
Sample type and preanalytical stability of FGF23
One major issue with FGF23 testing is that the sample requirements differ between assays. Immutopics recommend that only EDTA anticoagulated plasma should be used for FGF23 measurement, whereas according to Kainos, serum is the sample of choice. The reason for this discrepancy is unclear, but may relate to relative epitope stability in different sample matrices (e.g. divalent ion concentration). Our experiments using the Immutopics assay indicate that intact FGF23 is significantly more stable in plasma (with either lithium heparin or K2-EDTA anticoagulant) than in serum (unpublished data). A recent report by Fassbender et al. corroborates these observations and advocates that all FGF23 measurements should be made with plasma samples. 83 The use of EDTA plasma samples, however, does not allow simultaneous measurement of calcium concentration (which would be advantageous as a putative marker of bone mineral metabolism) and is dependent on adequate tube filling (>50%). In contrast, the use of serum delays testing to allow for clotting before centrifugation and processing, thus rendering impractical intraoperative venous sampling in patients with TIO.
The stability of the FGF23 was brought into focus in earlier studies by Imel et al. who noted that FGF23 concentrations were undetectable in a number of patients with histologically confirmed TIO using the Immutopics iFGF23 kit.
84
Similarly, using the same assay, Bacchetta et al. found that iFGF23 concentrations were undetectable in ‘historical sera’ from a series of patients with biopsy-proven TIO.
85
Whilst a small minority of tumours associated with TIO do not secrete FGF23, but rather release other phosphaturic factors (e.g. matrix extracellular phosphoglycoprotein and secreted frizzled-related protein-486), the most likely explanation for these findings is a loss of immunoreactive FGF23 due to degradation. Subsequently, we reported on the marked but variable instability of iFGF-23 measured using the Immutopics iFGF23 assay (ranging from 23.8 to 67.3% over 4 h),
71
which was significantly attenuated by the addition of a broad-spectrum protease inhibitor. Indeed, Immutopics acknowledge that ‘acceptable’ recovery (±10%) is only achieved with use of protease inhibitors. However, they provide no guidance on what protease protection strategy should be employed. The ex vivo proteolysis of plasma FGF23 is exemplified in the immunoblot shown in Figure 3, which depicts the loss of intact FGF23 immunoprecipitated from serum of a healthy volunteer, over a 48-h period following blood collection. At baseline the majority of FGF23 detection in this system is intact protein, but by 24 h postvenesection, the majority of the signal resides in the C-terminal fragment. Interestingly, however, instability appears to be less of an issue using Kainos and Millipore assays.
74
It is difficult to reconcile these findings completely, but may relate to differences in antibody specificity and the stability of particular FGF23 epitopes. Alternatively, the changes may reflect differences in the formulation of the assay buffer and addition of protease inhibitors (of note, the Millipore kit buffer contains proprietary protease inhibitors).
Instability of intact FGF23. Insta The instability of intact FGF23 protein.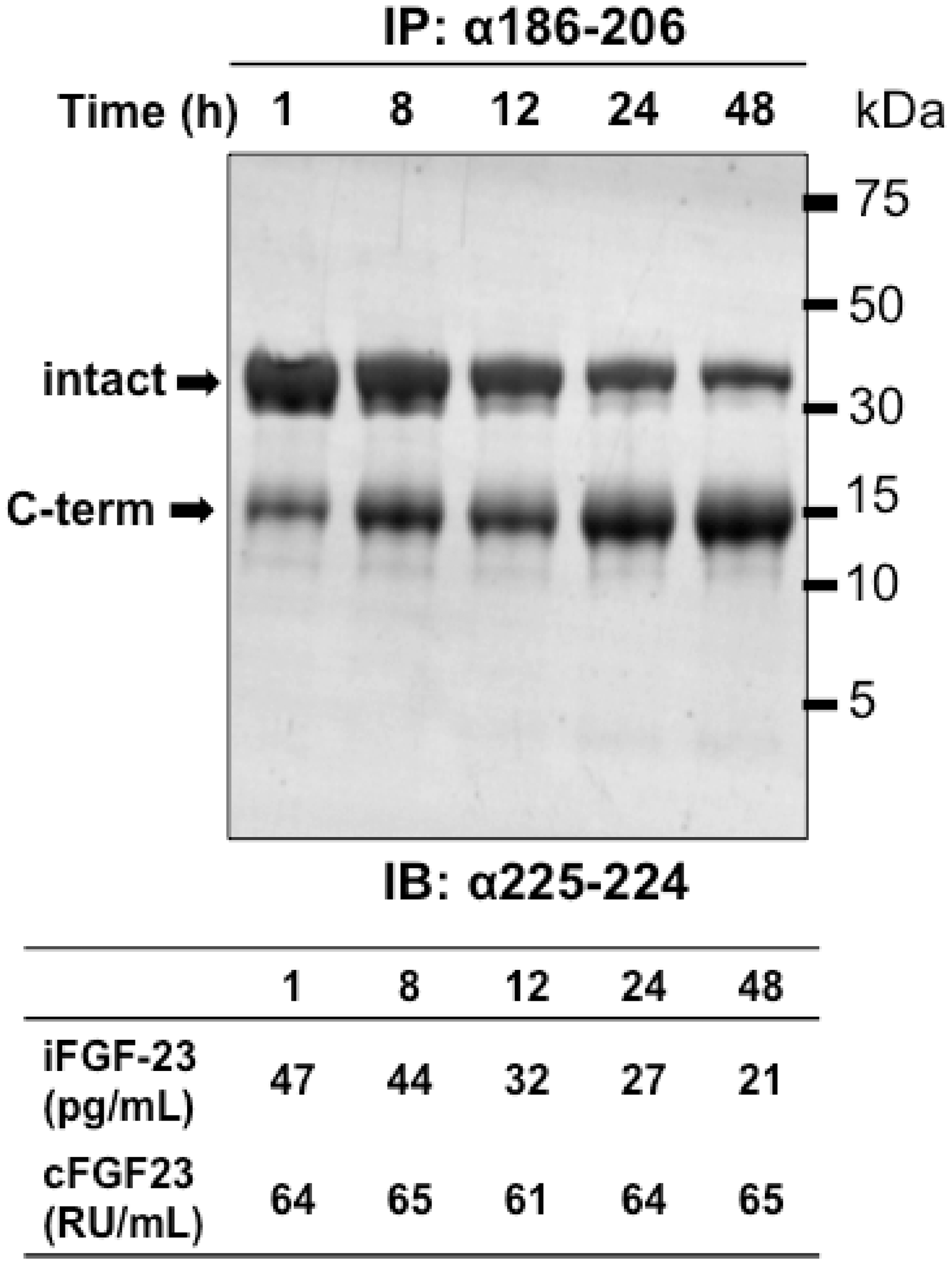
iFGF23 concentration is also affected by repeated freeze–thaw cycles, again affecting the Immutopics assay more than either Kainos or Millipore kits. Three freeze–thaw cycles (–20℃ to 24℃) were sufficient to cause significant loss of plasma iFGF23 immunoreactivity: mean 37, 11 and 15% decrease from baseline values for Immutopics, Kainos and Millipore assays, respectively (unpublished data). Thus, samples that have undergone repeated freeze–thaw cycles are unsuitable for analysis. Seemingly at odds with these findings, a recent study suggested that ‘pretest’ handling of samples has minimal effect on cFGF23 analysis, with measurements before and after storage and shipping (conditions not specified) being ‘highly correlated (>0.7)’. 41 Our study of FGF23 preanalytical instability also showed a highly significant correlation between baseline and 2-h time points after venesection (P = 0.001), but this belied the marked and variable changes in both iFGF23 (range ∼39–81% decrease) and first generation cFGF23 (range ∼18–75% increase) assays. 71 In our opinion, a correlation coefficient of only 0.7 in this context (same sample, same method) would indicate that at least a proportion of samples showed quite significant changes in FGF23 concentration due to pretest handling. In contrast to this, however, we have observed no evidence of significant instability in plasma FGF23 concentration using the second-generation Immutopics cFGF23 assay. 74
The apparent instability of intact FGF23 has led some investigators to favour the use of the cFGF23 assay, believing that the measurement of intact protein and c-terminal fragments ‘compensates’ for the effects of ex vivo degradation. This logic is flawed, however, as it overlooks the presence of endogenous, potentially counter-regulatory, fragments. 70 On theoretical grounds therefore, measurement of only iFGF23, once appropriately stabilized, would seem most desirable as this is likely to correlate more closely with biological effect. The use of blood collection tubes preloaded with inhibitor cocktail may prove to be the only way of reliably interpreting results by immunoassay, particularly within the physiological range, but such a strategy would be prohibitively expensive for widespread clinical use.
Biological variation
Plasma iFGF23 is subject to significant diurnal variation. Consistent with our recent report, 77 Vervloet et al. 87 showed that plasma iFGF23 concentrations peak in the early morning and fall during the day (mean decrease of 25%). This pattern might reflect changes in bone turnover (generally higher at night) and mirrors the fluctuations seen with other bone markers. Clearly, the diurnal variability of iFGF23 measurements has important implications for the timing of sample collection and assessment of therapeutic intervention. To evaluate the efficacy of FGF23-lowering therapy, early morning sampling appears preferable. In contrast, plasma cFGF23 concentrations show only a modest non-significant increase during the day (mean 8% increase), reaching peak values in the evening. The disparity in the variability of iFGF23 and cFGF23 concentrations is likely to reflect the differences in clearance of intact protein and C-terminal FGF23 fragment, 58 which might result in a somewhat blunted oscillation in cFGF23 concentration overall. Since FGF23 bioactivity may be indirectly regulated by phosphate intake, thought should be given to precisely when blood measurements are taken in relation to meal times, and their composition. However, there appears to be a substantial lag period (>12 h) in response to changes in dietary phosphate load;88,89 so recent consumption of food is unlikely to affect plasma concentrations significantly. Our data suggest that early morning (8:00–10:00) fasting plasma FGF23 concentrations, particularly iFGF23, are not significantly different from non-fasting concentrations. 77 Along similar lines, Isakova et al. report finding no significant postprandial changes in plasma cFGF23 concentrations in healthy adults or in patients with early CKD. 90
Compared to cFGF23 measurements, the inherent biologic variability of iFGF23 means that serial values have to change quite markedly for them to be considered statistically significant (reference change value, RCV ∼50% vs. 25%).
77
This also dictates the requirement for many more samples in order to accurately define an individual’s homeostatic set point (14 vs. 3 samples to be within ± 10% of the ‘true’ value). On the basis of these characteristics alone, the cFGF23 assay seems to have greater potential for clinical use. However, the low index of individuality for cFGF23 measurements (0.24) suggests that this test is not suited for use with conventional reference intervals;
91
as illustrated in Figure 4, an individual’s plasma concentration may change significantly for them but remain within the 95% population-based reference limits. Other clinical decision limits (risk- or therapy-based) may therefore be more appropriate, but have yet to be defined. Generally, analytes show greater variability in disease than in health,
92
as is the case for PTH in haemodialysis patients
76
and a number of bone turnover markers in Paget’s disease.
93
This generalization would also appear to apply to FGF23, since two recent studies report finding substantial intraindividual (week-to-week) variation in cFGF23 concentration in patients undergoing haemodialysis.94,95 Conversely, in other dialysis cohorts, the intraindividual variation of cFGF23 has been reported to be less than that of PTH.41,96 A caveat to these reports in patient populations, however, is the appropriateness (and hence reliability) of variance estimates based on parametric methods that assume the existence of steady state, in which terms are normally and independently distributed (i.e. absence of systematic changes during the study period) and in which variances are homogeneous.
97
In some of the aforementioned studies it does not appear that tests for outlier detection, variance homogeneity and linear trends have been performed to ensure these assumptions have been met. A further issue is that only one report gives confidence intervals of the derived estimates of intraindividual variability, greatly limiting direct comparison and interpretation. Notwithstanding this, the growing evidence base suggests that it would be unwise to base clinical decisions on any single FGF23 result in the management of an individual patient due to its inherent biological variability. Yet it should also be acknowledged that for the purposes of risk assessment (i.e. prognostication), cFGF23 measurements, in particular, demonstrate desirable variance characteristics, with much higher interindividual than intraindividual variation.
Biological variability of plasma FGF23 concentration using iFGF23 and cFGF23 assays. Week-to-week biological variation of non-fasting, early morning (08:00–10:00) plasma iFGF23 (left-hand side panel) and cFGF23 (right-hand side panel) concentrations in 12 healthy adult volunteers. Box plots show median (horizontal line), upper and lower quartile (box) and range (whiskers) of values for each participant. Dashed red lines indicate upper and lower 95% reference interval limits determined using respective assays. Reproduced and modified with permission from Ref. 77: http://jcem.endojournals.org.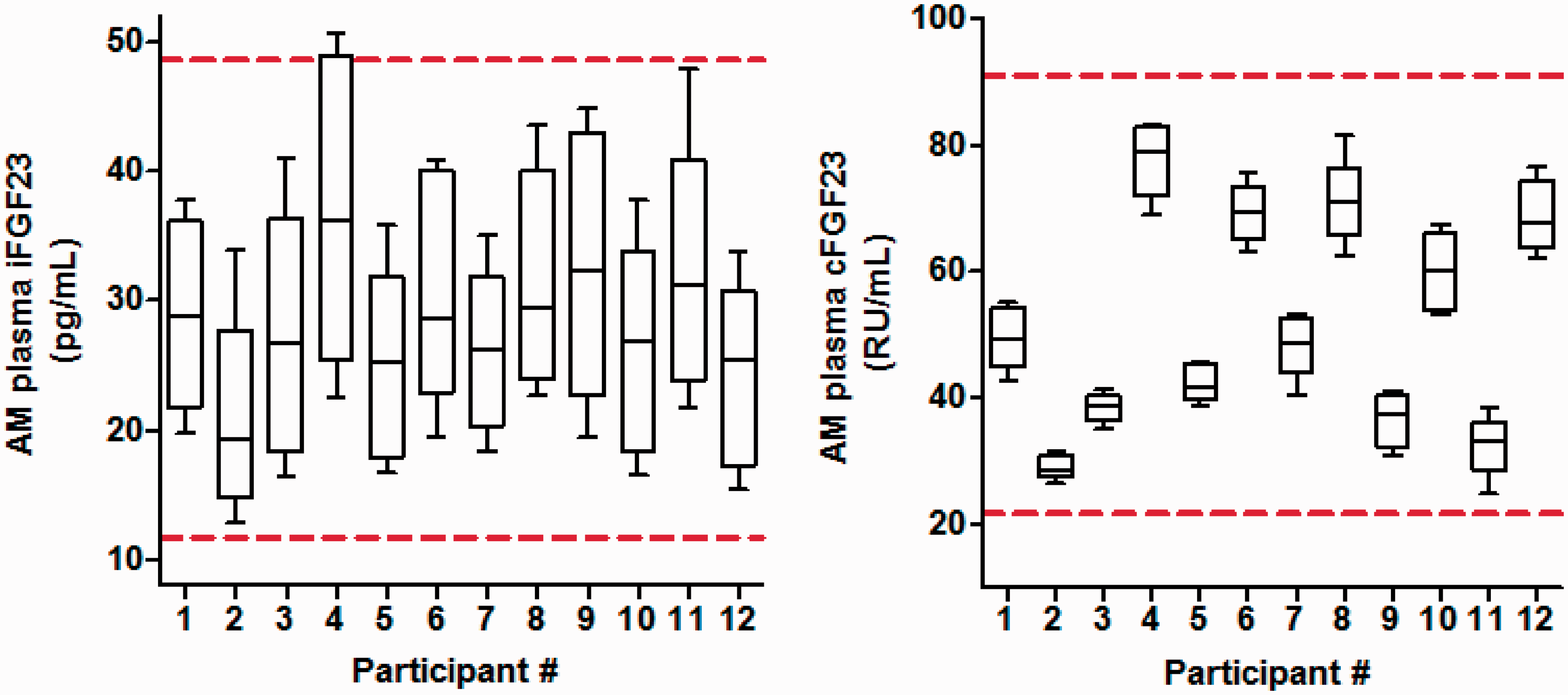
Accuracy and reproducibility of methods for FGF23 measurement
Assay and cohort-specific differences in analytical agreement
Lack of agreement between iFGF23 and cFGF23 ELISA assays in clinical studies.

Assay manufacturer: K, Kainos (Tokyo, Japan); I, Immutopics (San Clemente, USA); M, Millipore (Billerica, USA).
ADHR: autosomal dominant hypophosphataemic rickets; CKD: chronic kidney disease; FD: fibrous dysplasia of bone; HD: haemodialysis; LOD: analytical limit of detection; PD: peritoneal dialysis; TIO: tumour-induced osteomalacia; XLH: X-linked hypophosphataemic rickets; 1,25(OH)2D: 1,25-dihydroxyvitamin D.
Data comparing the different iFGF23 assays are scarce. In a study of the clinical sensitivity of plasma FGF23 measurements for diagnosis of TIO, Imel et al. found that the Kainos iFGF23 kit was superior (100% sensitivity) to either assay from Immutopics: cFGF23 (92%) and iFGF23 (32%). 99 We recently highlighted the very poor analytical agreement between iFGF23 assays from Kainos, Millipore and Immutopics, with the latter two assays yielding readouts very much lower than those measured with the Kainos kit. 74 Recovery experiments with full-length recombinant human FGF23 also indicated substantial under-recovery using the Immutopics assay, which was most pronounced at very high spiking concentrations. Differences in calibration mostly account for these discrepancies and underline the need for a common international standard preparation to allow harmonization of these assays.
Differential relationship of iFGF23 and cFGF23 with other parameters
Clinical studies in which cFGF23 and iFGF23 measurements show differing associations with biochemical and outcome variables.

Manufacturer: K, Kainos (Tokyo, Japan); I, Immutopics (San Clemente, USA).
AUC: area under curve; BMI: body mass index; CKD: chronic kidney disease; eGFR: estimated glomerular filtration rate; FCM: ferric carboxymaltose; HD: haemodialysis; HOMA-IR: homeostasis model assessment of insulin resistance; HR: hazard ratio; hs-cTnT: high-sensitivity cardiac troponin T; IGF-1: insulin-like growth factor-1; PD: peritoneal dialysis; PTH: parathyroid hormone; SCr: serum creatinine concentration.
Assay-dependent response of FGF23 to controlled-dietary phosphate intervention.

Manufacturer: K, Kainos (Tokyo, Japan); I, Immutopics (San Clemente, USA).
Al/Mg-OH: aluminium/magnesium hydroxide; AUC: area under curve; Ca: calcium; CKD: chronic kidney disease; HCl: hydrochloride; HD: haemodialysis; L: loading diet; P: phosphate; R: restricted diet.
Endogenous and pharmacological modulators of plasma FGF23 concentrations – factors to consider when interpreting results.

Differential effect on iFGF23 and cFGF23 concentrations.
FGFR1: fibroblast growth factor receptor 1; IL-6: interleukin-6; NC: no change; VDRA: vitamin D receptor activators.
Elimination studies – utility of FGF23 analysis for tumour localization and monitoring in patients with TIO
Further evidence of dissimilarity comes from studies of the clearance of FGF23 from plasma after tumour resection in patients with TIO. The half-life of FGF23 differs substantially depending on whether a cFGF23 or iFGF23 assay is used. 112 Khosravi et al. found that plasma FGF23 concentrations generally decayed more slowly when measured with the Immutopics cFGF23 assay (t1/2 58 ± 34 min) than with the Kainos iFGF23 assay (t1/2 46 ± 12 min) and remained elevated beyond 72 h compared to iFGF23 concentrations, which became mostly undetectable within 24 h. This is consistent with a more recent study of patients with TIO who underwent serial monitoring of FGF23 concentrations after tumour removal. 113 Here, iFGF23 concentrations were found to drop rapidly to almost undetectable levels within hours of resection, but returned to normal within 5 days; while cFGF23 concentrations fell at a slower rate but remained elevated for the 10-day follow-up period. This suggests that C-terminal fragments may be cleared less rapidly than intact hormone and is consistent with elimination studies previously conducted in rats. 70 In terms of the utility of FGF23 measurements for selective venous sampling procedures and assessment of biochemical remission following resection, it also suggests that iFGF23 analysis is likely to be more sensitive than cFGF23 readout. The fact that iFGF23 changes so quickly after removal of a culprit lesion suggests that intra- and postoperative measurement may be a potential tool to assess the success of surgery, akin to PTH sampling during parathyroidectomy.
FGF23 fragments – to be or not to be?
Early work suggested that C-terminal FGF23 fragments were present in the serum of healthy adults, but were particularly abundant in serum from ESRD patients. 114 In the context of CKD, therefore, it was initially theorized that the high concentration of circulating FGF23 was due to reduced renal clearance and accumulation of C-terminal fragments. However, this appeared not to be the case when Shimada and colleagues subsequently demonstrated that the FGF23 present in the plasma of patients with ESRD undergoing peritoneal dialysis was almost exclusively full-length intact hormone. 81 Consistent with Shimada et al., we also found intact iFGF23 to be the predominant species in the plasma of patients undergoing haemodialysis. 77 However, in contrast to these findings, C-terminal FGF23 fragments were present in plasma of healthy adults and, in lesser amounts, in patients with predialysis CKD. 77 Thus, it would seem that as eGFR declines the fraction of FGF23 present as intact hormone increases, which may well explain why cFGF23 and iFGF23 measurements show closer agreement in patients undergoing dialysis than in other populations. Indeed, even in settings where iFGF23 and cFGF23 are strongly correlated (e.g. in patients on dialysis), plasma FGF23 concentrations (determined using the iFGF23 assay) show a much more convincing relationship with FGF23 bioactivity (estimated using a HEK cell-based Erg-1 luciferase reporter assay) than concentrations ascertained with the cFGF23 kit (R2 = 0.93 vs. 0.74, respectively). 81 Hence, FGF23 measurement using the cFGF23 assay does not necessary reflect iFGF23 bioactivity but instead is a composite measure of two antagonistic species.
That the ratio of iFGF23 to fragments might increase as renal function worsens may seem counterintuitive: other hormone fragments tend to accumulate in CKD due to reduced clearance. Furthermore, in patients with TIO, cFGF23 concentrations tend to a longer half-life in plasma than iFGF23.69,113 Accounting for these findings is challenging. One hypothesis would be that as renal function fails, the increasing stimuli for FGF23 production (dysregulated bone mineralization, phosphate retention, end-organ resistance/α-klotho deficiency, iron deficiency, hyperparathyroidism, etc.) may overwhelm the capacity of the physiological inactivation mechanisms that ordinarily keep bioactive FGF23 concentrations in check. Alternatively, proteolytic machinery may be down-regulated by the uraemic milieu. Further work is needed to explore these concepts and evidence is needed to confirm the existence and function of C-terminal FGF23 fragments.
Diseases associated with disturbances in FGF23 processing
Several diseases have been associated with disrupted processing of FGF23 in bone. In these instances there can be very marked differences in iFGF23 and cFGF23 assay readout. Destabilizing mutations in FGF23 (including missense mutations at glycosylation sites) or inactivating mutations in GALNT3 result in increased proteolysis and loss of its phosphaturic effects. 115 This is seen in patients with the hyperphosphataemic hyperostosis syndrome or tumoral calcinosis, diseases characterized by severe hyperphosphataemia, episodic bone pain, localized cortical hyperostosis and ectopic calcification. A similar phenotype is seen in patients with loss of function mutations in α-klotho, leading to end-organ resistance. 116
Patients with fibrous dysplasia (FD) of bone also often have elevated plasma FGF23 concentrations, yet the classical phosphate-wasting phenotype as seen in other hypophosphataemic syndromes of FGF23 excess is relatively uncommon in this disorder. 117 Significantly, Bhattacharyya et al. recently reported higher ratios of cFGF23 to iFGF23 in patients with FD compared to healthy controls (mean 1.7 vs. 0.99). 98 The authors also provided primary evidence of modified FGF23 processing from analysis of bone marrow stromal cells harbouring the causative activating Gsα mutation (affecting the α-subunit of the stimulatory G-protein), which showed a cAMP-dependent inhibition of GALNT3 activity and increased furin activity. Conversely, cFGF23 to iFGF23 ratios in patients with renal failure were highly variable, ranging from ∼0.1 to ∼4 RU/pg. Such observations, especially ratios <1, are difficult to interpret, but are likely to reflect a combination of analytical imprecision and differences in calibration. To facilitate more meaningful interpretation of ratio data it would be sensible to calibrate both assays with the same set of standards. Nonetheless, in situations where altered FGF23 processing may be suspected, it would seem prudent to consider measurement of FGF23 using iFGF23 and cFGF23 assays simultaneously.
FGF23 – the new PTH or partners in confusion?
Over the last few years, much has been said about the suitability of PTH measurements for use in nephrological practice, and, despite reliance on it for several decades to guide management of bone disease in CKD patients, some critics now question its continued use altogether. 118 The pitfalls of PTH measurement are all too familiar: poor agreement between assays (due partly to differences in calibration but also to the variable detection of PTH fragments), substantial diurnal and day-to-day intraindividual biological variability and the lack of a universal standard. 118 This has prompted many investigators to search for a ‘better’ alternative to PTH-based testing protocols for the diagnosis and management of CKD-MBD. In this context, FGF23 measurement has been put forward as a possible substitute for PTH,6,41 but is there evidence to support this claim?
Plasma FGF23 concentrations increase early in CKD, probably preceding increases in PTH concentration. 6 Although consistent with animal studies, 11 the sequence of biochemical changes in human CKD is a matter of debate, as the available evidence is based on single-point cross-sectional determinations and fails to take into account differences in the analytical and biological variability of plasma cFGF23 and PTH measurements. Indeed, in health, the intraindividual variation in plasma PTH concentration (19.2–25.9%) is an order of magnitude greater than that of plasma C-terminal FGF23 (6.9%).77,119 A longitudinal study is needed to properly define the temporal relationship between changes in FGF23 and PTH. On the other hand, in haemodialysis patients, Cavalier et al. report that cFGF23 has a higher intraindividual biological variability than any other measured marker of bone mineral metabolism, including contemporary second- and third-generation PTH assays. 94 With respect to biological variability, therefore, the choice of assay or indeed biomarker is not straightforward. Instead, the data perhaps suggest that highly dynamic, acutely regulated hormones like PTH or FGF23 are not a good clinical target for diagnosis or therapeutic monitoring.
It is important to emphasize however that PTH and FGF23 measurements clearly do not carry the same information. According to a study by Isakova et al., 6 only a proportion of patients with mild-to-moderate CKD (33% eGFR ≥ 70 mL/min/1.73 m2; >50% eGFR 50–59 mL/min/1.73 m2) have elevated plasma cFGF23 concentrations (≥100 RU/mL). Similarly, only a proportion of these patients had raised PTH concentrations and, perhaps most significantly, not all patients with increased plasma PTH concentrations had elevated plasma cFGF23 concentrations. In part, this may reflect the inappropriateness of interpreting cFGF23 with respect to ‘normal’ ranges due to its high index of individuality, but it also highlights that measurement of either FGF23 or PTH alone may not necessarily capture the full spectrum and complexity entailed within the diagnosis of CKD-MBD. A combinatorial approach based on a number of markers may therefore yield better clinical sensitivity, but this has yet to be demonstrated.
Comparison between preanalytical and analytical aspects of FGF23 and PTH measurements.
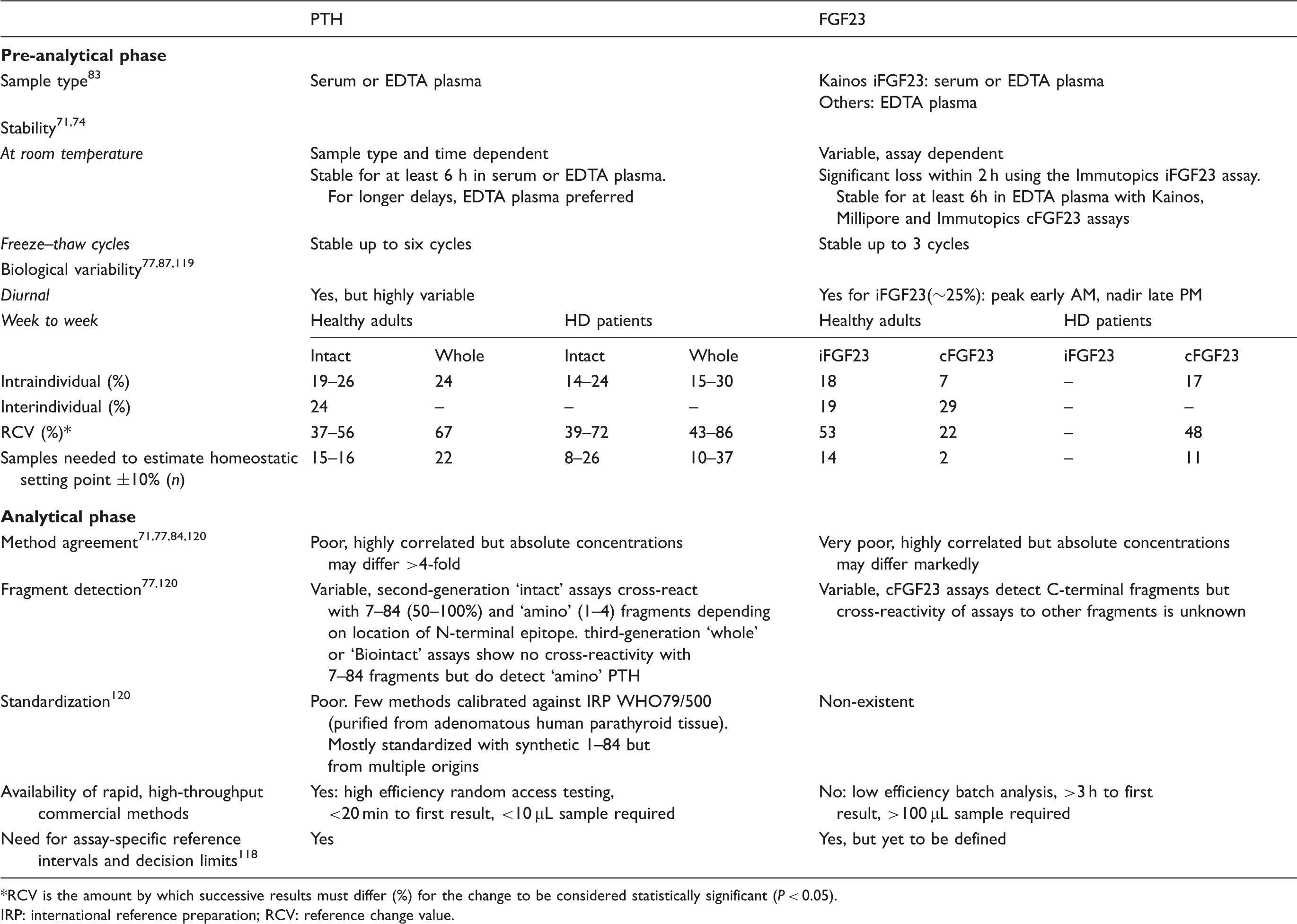
RCV is the amount by which successive results must differ (%) for the change to be considered statistically significant (P < 0.05).
IRP: international reference preparation; RCV: reference change value.
While it is certainly the case that FGF23 concentration appears much more strongly and robustly associated with hard outcomes in CKD, (and in the wider community) than PTH, 123 these epidemiological findings do not necessarily translate into improved clinical performance. In some respects, the similar difficulties faced when testing either hormone strongly suggests caution before proposing that routine testing of FGF23 (by whatever assay) should be integrated into clinical practice. More work is needed to examine its clinical credentials and suitability for ‘real-world’ use: decision limits have yet to be defined and validated; clear evidence that FGF23 measurement adds to or improves risk assessment in the clinical setting; and, ideally, evidence that modification of FGF23-associated risk or FGF23-guided monitoring improves patient outcome.
Concluding remarks – state of the art or state of uncertainty?
FGF23 has emerged as a promising biomarker for predicting adverse outcomes in patients with CKD. If it can be shown that therapies aimed at reducing FGF23 bioactivity have benefit, then routine measurement of this hormone may prove useful. However, before serious consideration can be given to its potential clinical utility, various preanalytical and analytical issues need to be thoroughly addressed. Due to differences in fragment detection, antibody specificity and calibration, the current ‘state-of-the-art’ ELISA methods do not permit direct comparison of FGF23 measurements, particularly outside the ESRD setting. From a clinical utility point of view, these differences also thwart the creation of standardized reference intervals or clinical decision limits that are central to patient management guidelines. Moreover, these methods are not suited to the modern high-throughput laboratory, where rapid, sensitive and sample volume-sparing analyses are the mainstay. Finally, the variable instability of intact FGF23 necessitates careful and judicious sample collection to preserve the in vivo protein state and to gain an accurate assessment of FGF23 bioactivity. Undoubtedly, these factors have together contributed to the inconsistency and variability in the FGF23 literature and may equally impact negatively on their use in clinical practice.
Critically, more work is needed to (1) standardize and harmonize the output of commercial assays, (2) develop a human (native) plasma-based or appropriate synthetic reference standard, (3) determine and validate assay-specific reference intervals and/or decision limits, (4) collect more data on the cleavage of FGF23 and the biological effects of fragments, (5) determine how analysis could be incorporated into CKD management strategies and evaluate whether such an approach is both beneficial and cost-effective for the patient.
Review criteria
PubMed was searched for articles published up to 12 July 2013 using the search terms ‘fibroblast growth factor 23’, ‘FGF-23’, ‘FGF23’, ‘phosphatonin’. All English language, full-text articles were evaluated for their inclusion in the review. Reference lists of publications were also examined to identify additional relevant articles.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of Competing Interests
None.
Funding
ERS, LPM and SGH have received research funding from Amgen and Baxter. ERS has received honoraria from Shire; LPM has received honoraria from Amgen and Roche; SGH has received honoraria from Amgen, Baxter, Gilead and Shire.
Ethical approval
None.
Guarantor
ERS.
Contributorship
ERS wrote the review. LPM and SGH commented on the draft.