
Abstract
Background
Neonatal screening for haemoglobin (Hb) disorders is a standard of care in several developed countries with the main objective to detect Hb S. Such practice has not been established in Thailand where α-thalassaemia and haemoglobin E (Hb E) are highly prevalent. Early identification of thalassaemias could be helpful and strengthen the programme for prevention and control for severe thalassaemias.
Methods
Data from isoelectric focusing (IEF) and Isoscan® for detecting types and amount (%) of each haemoglobin in 350 newborn’s dried blood spots were analysed and compared with the comprehensive genotype analysis by DNA studies as a gold standard.
Results
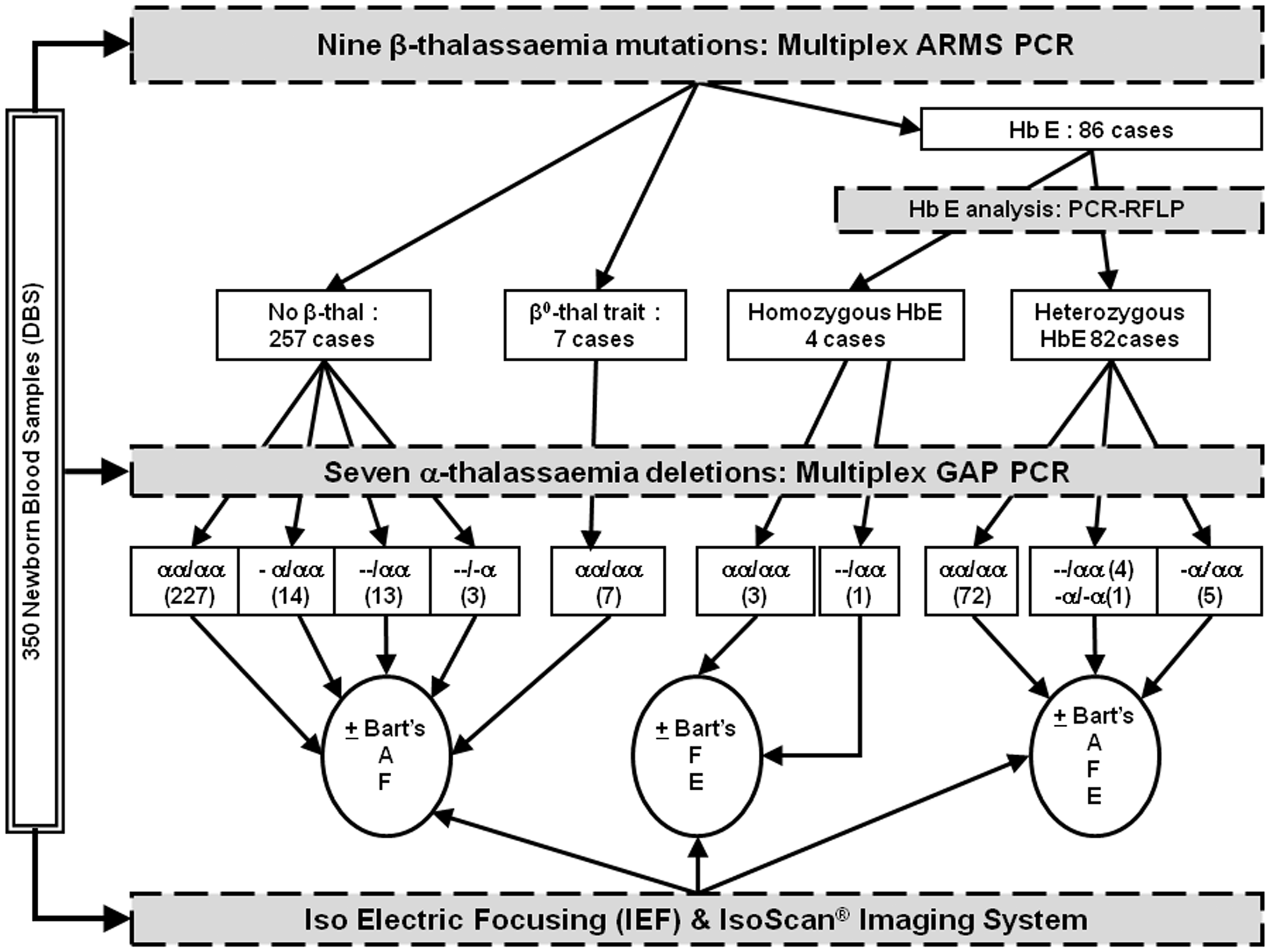
Based on genetic profiles, there were 10 different categories: (1) normal (n = 227), (2) α+-thalassaemia trait (n = 14), (3) α0-thalassaemia trait (n = 13), (4) β0-thalassaemia trait (n = 7), (5) Hb E trait (n = 72), (6) Hb E trait with α0-thalassaemia or homozygous α+-thalassaemia (n = 5), (7) Hb E trait with α+-thalassaemia trait (n = 5), (8) homozygous Hb E (n = 3), (9) homozygous Hb E with α0-thalassaemia trait (n = 1) and (10) Hb H disease (n = 3). The presence of Hb Bart’s and Hb E were used to identify cases with α-thalassaemia and Hb E, respectively. We set 0.25% of Hb Bart’s and 1.5% of Hb E as a cut-off level to detect α+-thalassaemia trait (sensitivity 92.86% and specificity 74.0%) and Hb E trait with 100% of both sensitivity and specificity for IEF diagnosis.
Conclusion
Although molecular diagnosis seems to be better for definitive diagnosis of thalassaemia syndromes at birth, however, using our reference range described herein, IEF can be applied in a resource-limiting setting with acceptable reliability.
Keywords
Introduction
Thalassaemia and haemoglobinopathies, characterized by a decrease and abnormal production of globin chains, are common genetic blood disease and a major public health problem in many countries worldwide including Thailand. 1 The programme for thalassaemia prevention and control composed of a prenatal screening by mean red blood cell volume or osmotic fragility (OF) in combination with Hb E detection by dichlorophenol indophenol (DCIP) dye in pregnant women has been established in Thailand since 1994. The screening scheme aims to identify couples at risk of β-thalassaemia major (β0/β0), Hb E/β thalassaemia (βE/β) and Hb Bart’s hydrops fetalis (- -/- -); however, the outcomes have not been widely successful. 2 One of the main drawbacks was couples at risk do not have enough knowledge and awareness about their conditions and possible consequences. Moreover, although more than 800,000 pregnant women have successfully been screened through the programme, only 20–60% of their male partners were willing to be tested. 3 Therefore, the effectiveness of detecting a couple with a reproductive risk for thalassaemia was hampered and less than 10% of expected fetus with severe thalassaemia syndromes was prenatally diagnosed each year. 2
Due to a high frequency of the α and β thalassaemia genes in Thailand, approximately 40% of our population is thalassaemia carriers. Nearly 20% of this group is double heterozygotes for both globin mutations. 3 For α-thalassaemias, the majority of carriers are α+-thalassaemia by one gene lost either 3.7- or 4.2-type deletions (-α3.7/αα or -α4.2/αα) followed by α0-thalassaemia by deletions of two linked α-globin genes. The SEA type of α0-thalassaemia (- -SEA/αα) is the most common deletion accounted for 99% of cases identified. 4 In addition, α+-thalassaemia can result from non-deletional mutations (αTα/αα or ααT/αα) and two termination codon mutations; Hb Constant Spring (HBA2:c.427T > C) and Hb Paksé (HBA2:c.429A > T) were found in most cases 5 although several missense and nonsense mutations are increasingly identified. 6 For β-globin genes, more than 30 different β-globin mutations have been found together with several β-haemoglobinopathies (variants). The most common and important variant is Hb E due to A→G mutation at codon 26 (HBB:c.79G > A). 7 These complex interactions of both globin mutations are troublesome on making definitive diagnosis even at childhood and adult period. 8 Therefore, it is challenging for Thailand and possibly other Southeast Asian countries where both α and β-globin mutations are frequent to accurately set a screening cut-off for clinical diagnosis of different thalassaemia genotypes since the amounts of haemoglobin of interest might be significantly reduced due to interaction of another globin locus. 1 Therefore, it is of important for any validation and optimization study for thalassaemia and haemoglobinopathy screening in Thailand to comprehensively analyse using molecular testing as a ‘gold’ standard to compare with proposed screening model.
A programme for newborn screening has started in Thailand since 1994 at Siriraj Hospital in Bangkok to identify two common causes of mental retardation; congenital hypothyroid (CH) and phenylketonurea (PKU) and subsequently adopted nationwide in 1996.9–12 The programme uses a dry blood spot on a filter paper or ‘‘Guthrie card’’ taken from a heel-prick of newborns within 24 h after delivery and measure serum markers at central laboratories.10,11 The coverage of our current newborn screening programme was nearly 100% of the babies born in our health care centres in the country. 10 To this regard, a programme to identify clinically significant thalassaemia syndromes and carriers at the neonatal period might be of helpful to improve the effectiveness on our current national prevention and control programme. In addition, such newborn screening programme for haemoglobinopathy should be economically feasible and could simply be applied into our current practice.
In this study, we evaluated the use of haemoglobin analysis on dry blood spots from filter papers in 350 newborns by isoelectric focusing (IEF) followed by quantitation of haemoglobin bands by Isoscan® Image System. These haemoglobin data were analysed in comparison with comprehensive globin genotypes that provided a definite diagnosis in each individual. A standard reference range of each genotype was set and tested for their sensitivity and specificity. This strategy can fit perfectly well with our current newborn screening program. The main objective of the study is to test whether newborn screening programme for haemoglobinopathy can provide a novel model to identify individuals with clinically significant thalassaemias and globin disorders in our population. Such strategy might be useful to improve a long-term success on our prevention and control programme for severe thalassaemia syndromes.
Materials and methods
Sample materials
Total 350 newborns who were delivered from 1 April 2008 to 31 August 2008 at delivery room, Department of Obstetrics & Gynecology, Phramongkutklao Hospital have been recruited into the study after the informed consent was obtained prior to the study from their parents. Their blood samples were collected on filter papers as a part of the routine care for neonatal screening of CH and PKU. A total of four dried blood spots were used for this study. One spot was for haemoglobin analysed using IEF technique and the others three spots were used for DNA analyses. The study proposal was submitted to the Ramathibodi and Phramongkutklao Hospital Ethic Committee (No.2008/1051) and has been approved by the committee on human rights related to research studies involving human subjects, based on the Declaration of Helsinki. All specimens were stored at −20℃ within 6 h after collection and kept until haemoglobin analysis within 7 days.
Haemoglobin analysis by isoelectric focusing
The paper disc was placed in a sample cup and 25 µL of Hb elution solution was added. The sample with the Hb elution solution was mixed well and kept for at least 30 min or until the haemoglobins were completely eluted from the paper disc. All haemolysate samples were applied to a standard IEF gel using RESOLVE® testing equipment and RESOLVE® Hemoglobin and Neonatal Hemoglobin kits. Isoscan® Imaging System was used to evaluate percentage and isoelectric point (pI) of all detected haemoglobins and the quantitation of bands’ density was performed according to the manufacturers’ instruction (Perkin Elmer Life Sciences, Zaventem, Belgium). To test whether the stability of haemoglobins remain conserved for routine service, we stored the blood spot samples at −20℃ before IEF and image scan at different durations. We determined the effect of storage time on the concentrations of several haemoglobins identified especially Hb E and Hb Bart’s. Five newborn dried blood spot samples that were examined for this analysis were stored for 1, 7, 14 and 21 days.
Molecular analyses of common α and β thalassaemia genes in Thailand
Genomic DNA was extracted from dried blood spots using salting out method. All samples were tested with an extensive combined set of polymerase chain reaction (PCR)-based methods to detect common α and β thalassaemia mutations found in Thailand; a set of GAP-PCR analysis to identify seven α-thalassaemia deletions; - -SEA, - -THAI, - -FIL, - -MED, - -(20.5), -α3.7 and -α 4.2 as described. 13 A single tube multiplex amplification refractory mutation system (ARMS)-PCR analysis was performed to detect six non-deletional α-thalassaemia mutations in Thais including; an initiation codon mutation (ATG > A–G; HBA2:c.2delT), α2 codon 30 GAG deletion (HBA2:c.91_93delGAG), Hb Adana (HBA2 or HBA1:c.179G > A), Hb Quong Sze (HBA2:c.377T > C), Hb Constant Spring (HBA2:c.427T > C) and Hb Paksé (HBA2:c.429A > T). These molecular two tests can cover >98% of α-thalassaemia alleles found in Thailand. 6 For mutations in the β globin locus, we used a newly devised single-tube multiplex ARMS-PCR to detect nine common β -thalassaemia mutations in Thailand: –28(A–G)/HBB:c.–78A > G; codon 8/9 (+G)/HBB:c.27_28insG; codon 17 (A–T)/HBB:c.52A > T; codon 26 (A–G or Hb E/HBB:c.79G > A); IVS–I–1 (G–T)/HBB:c.92 + 1G > T; IVS–I–5 (G–C)/ HBB:c.92 + 5G > C; codon 41/42 (–TCTT)/HBB:c.126_129delCTTT; codon 71/72 (+A)/HBB:c.216_217insA and IVS–II–654(C–T)/HBB:c.316–197C > T. Identification of these clinically significant mutations could account for >95% of β-thalassaemia alleles found in our centre. 7 All primers and conditions for these two ARMS-PCR tests are available upon request. All samples in which were identified with Hb E alleles by multiplex ARMS-PCR, they were further analysed by using PCR-RFLP using Mnl I digestion to discriminate between homozygotes and heterozygotes of Hb E mutation as described previously. 14 Individuals with unusual increased Hb Bart’s, but were not positive for common α-thalassaemia alleles, would be directly sequenced of the α2 and α1 globin genes using standard techniques. 5
Statistical analysis
All haemoglobin data together with definitive molecular results were analysed to identify a cut-off value for each globin genotypes. SPSS statistical software package Version 16.0 (SPSS Inc, 2000) was used for data analysis of this study. Receiver-operating-characteristic (ROC) curve was used to determine the Hb Bart’s cut-off level for diagnosis of α+-thalassaemia carrier. Independent samples t-tests were used to determine the difference between groups. Two ×two table tests were performed to determine sensitivity and specificity of IEF technique and DNA analysis was used as gold standard for all samples tested.
Results
Summary of all globin genotypes and haemoglobin profiles in 350 newborns
The IEF patterns on screen of Isoscan® programme of all genotype groups analysed are shown in Figure 1. Normal newborns produce approximately 60–85% fetal haemoglobin (Hb F, pI 7.0), 15–40% adult haemoglobin (Hb A, pI 6.90) and little or no Hb A2 (L1 = Lane 1 in Figure 1). Hb F was partially acetylated and identified as an Hb band moved to a more cathodic position to Hb F (Hb Fac) and comprising about 10% of the total Hb F. One hundred and thirty-one cases (37.43%) were observed in this profile and they were confirmed by normal α and β globin genotypes by DNA study. A large proportion of newborns studied were found to present with a slow moving band (pI 7.60) in a position suggesting Hb A2 or Hb E. However due to a normal Hb switching process, Hb A2 is less expressed at birth, therefore such detected Hb band was identified as Hb E.
15
In addition, infants who were detected Hb Bart’s as a fast moving band located at pI range from 6.0 to 6.40, suggesting α-thalassaemia.16–18 There were six patterns of the haemoglobin profiles that were detected comprising FA (±Bart’s), EFA (±Bart’s) and EF (±Bart’s). These findings were correlated independently with the molecular findings showing 10 different α and β globin genotypes as shown in Figure 2. Three hundred and six newborns (87%) had normal α-globin alleles (αα/αα). Ninety-three individuals were identified with β globin mutations (26%): four homozygotes Hb E (βE/βE, 1.14%), 82 Hb E trait (βE/β, 23.42%), five codon 41/42 traits (β41/42/β, 1.43%) and two codon 17 traits (β
17
/β, 0.57%).
Iso-electric focusing patterns of 10 genotypes analysed by Isoscan®. Summary of diagnostic scheme performed in 350 dry blood spot samples from Thailand newborn screening. DBS = dry blood spot.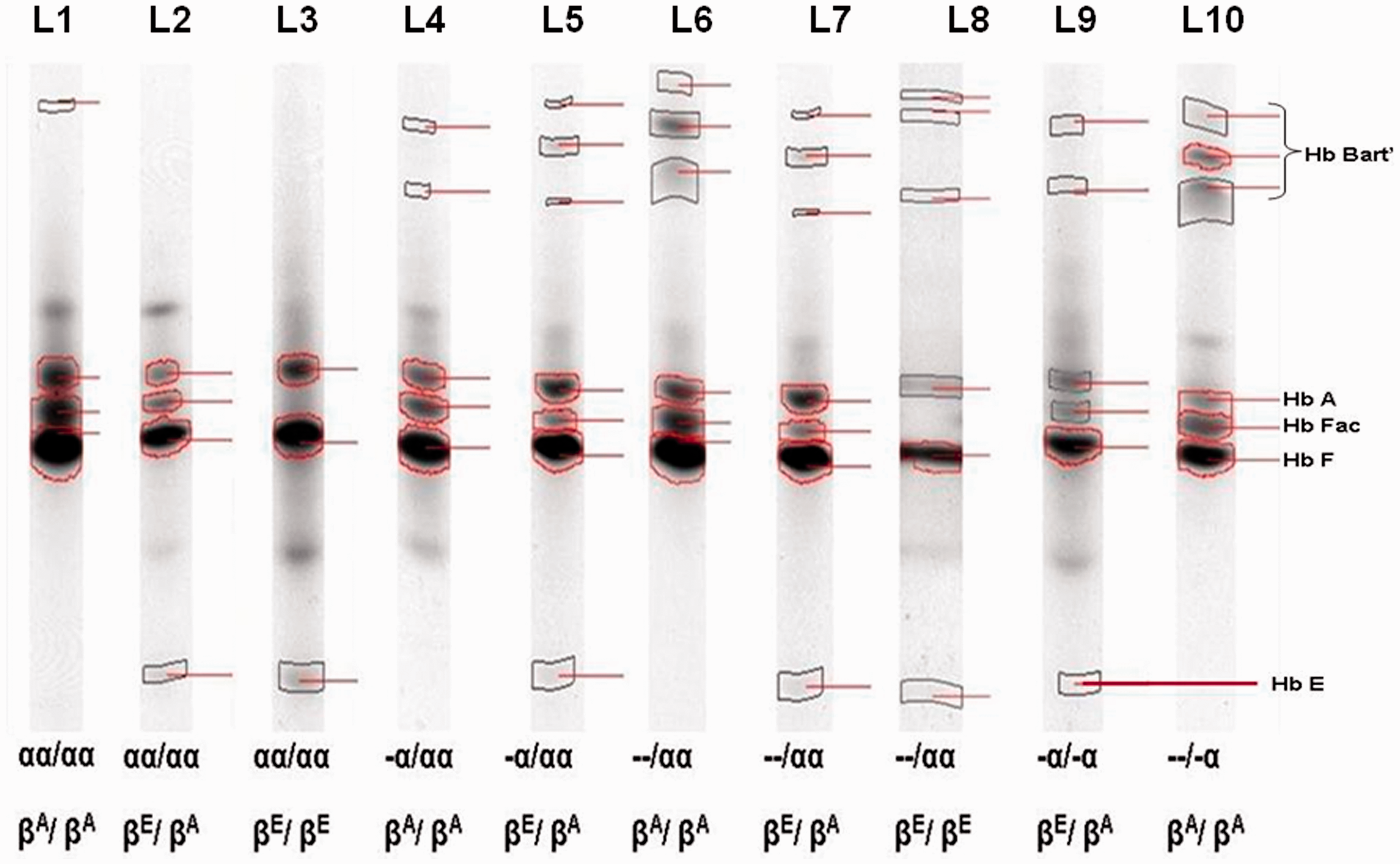
Measurement of Hb Bart’s and haemoglobin E in different genotype groups
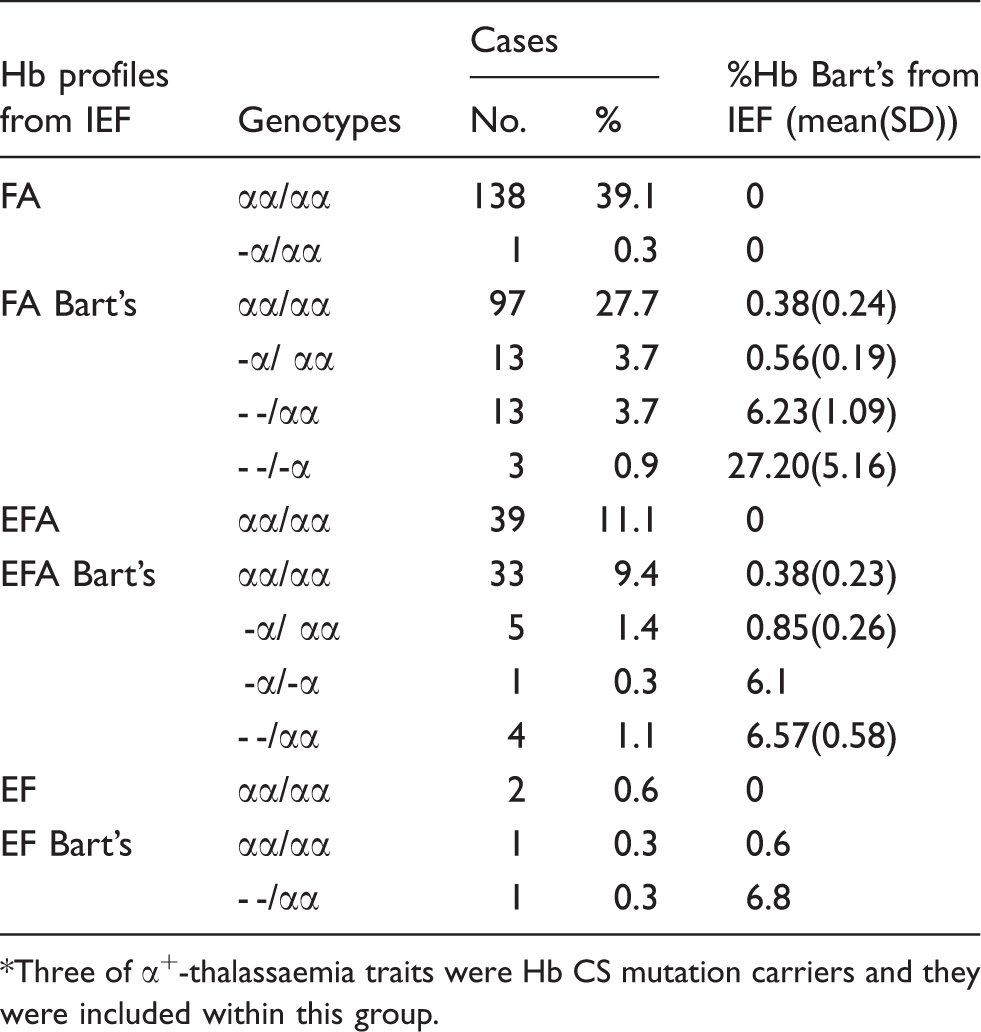
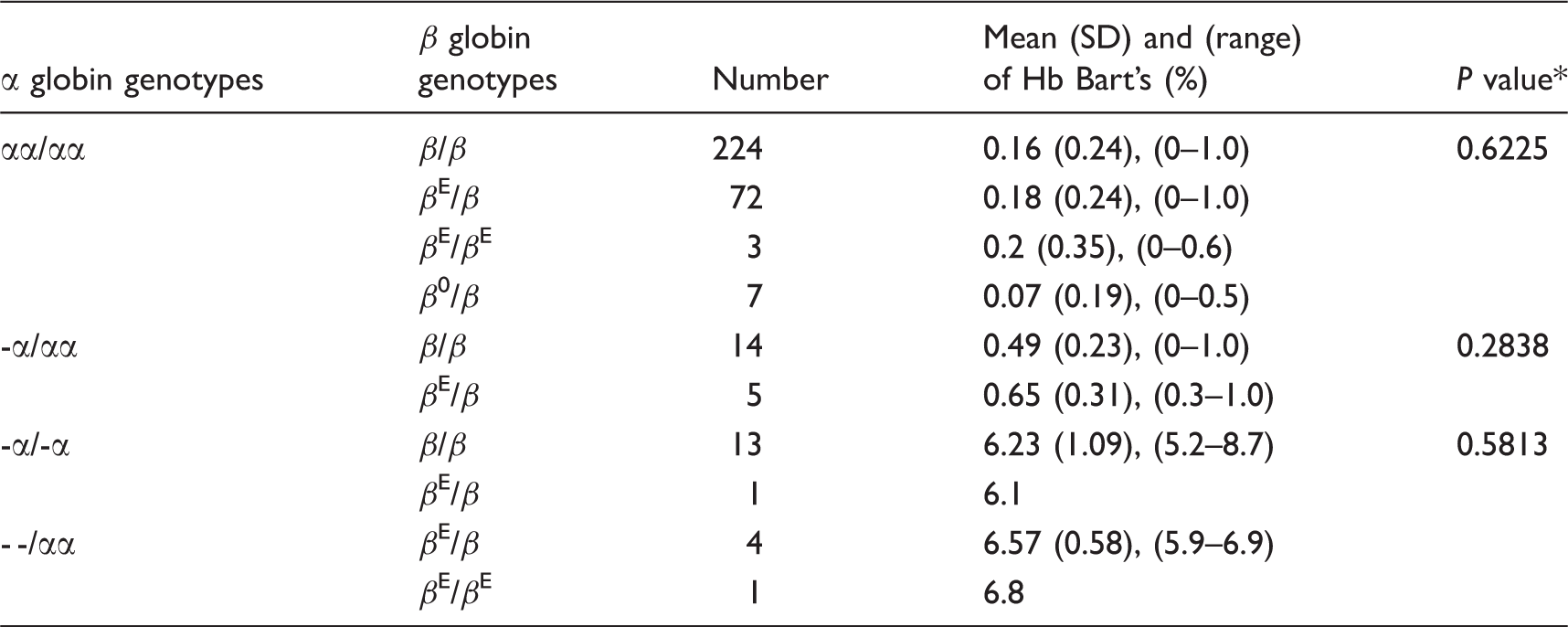
Hb Bart’s was found in 168 cases (48.3%) and ranged from 0.1% to 33.0% of total Hbs. The others 182 cases (51.7%) had not been detected with Hb Bart’s and all of them were negative for α-thalassaemia detections by GAP and ARMS-PCR analyses. The small amount of Hb Bart’s (mean (SD); 0.38 ± 0.24%) was detected in newborns with intact α globin genotype (n = 96, 27.43%). Three out of 22 heterozygotes for α+-thalassaemia were in fact Hb CS traits (αCSα/αα), whereas six were 4.2 kb-deletion (-α4.2/αα), and the rest were 3.7 kb-deletion type (-α3.7/αα). All heterozygous for -α3.7 or -α4.2 deletion (n = 19) had Hb Bart’s in the range of 0 to less than 1% which was overlapping with 306 individuals with normal α globin genotype (αα/αα) while Hb CS traits had Hb Bart’s at 1.0%, 1.1% and 1.5%. Since there was no different of Hb Bart’s concentrations among different α+-thalassaemia genotypes, they were all included together for the further analyses. Three cases of Hb H disease were compound heterozygotes for SEA-type and 3.7 kb-deletions (- -SEA/-α3.7; mean Hb Bart’s; 27.2 ± 5.16%) and all α0-thalassaemia traits carried the SEA-type deletion (- -SEA/αα, n = 18). One homozygous 3.7 kb-deletion was identified (-α3.7/-α3.7) with 6.1% Hb Bart’s and this case was further included with the group of α0-thalassaemia trait (with mean Hb Bart’s; 6.23 ± 1.09%). All α globin genotype and Hb Bart’s concentrations (mean (SD)) are summarized in Table 1 and Figure 3. In addition, 128 out of 350 samples were detected with Hb A2/E. However, only 86 cases of them, with the concentrations ranging from 1.5% to 4.5%, were confirmed as being heterozygotes for Hb E by DNA analysis. Forty-two cases detected with a quantity of <0.6% of total haemoglobin were negative for Hb E mutation. Six individuals with homozygous Hb E had the concentrations of Hb E of 4.8–5.6%.
Concentrations of Hb Bart’s and Hb E in cases with different α thalassaemia and Hb E genotypes. Six categories of Hb profiles in association with genotypes and the concentrations of Hb Bart’s in 350 Thai newborns. *Three of α+-thalassaemia traits were Hb CS mutation carriers and they were included within this group.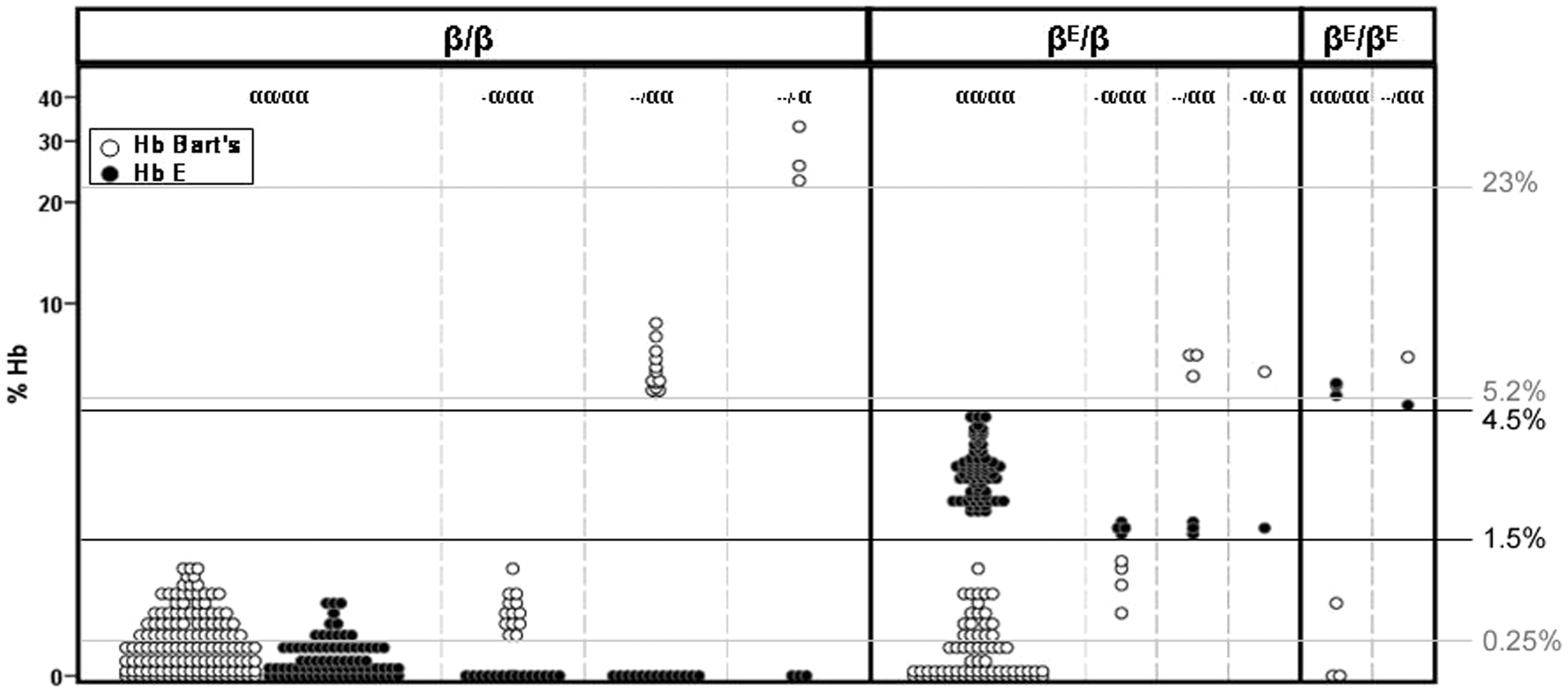
Effects of α-globin genotypes on the concentrations of haemoglobin E at neonatal period
Effects of α-thalassaemias on the concentrations of Hb E at neonatal period.
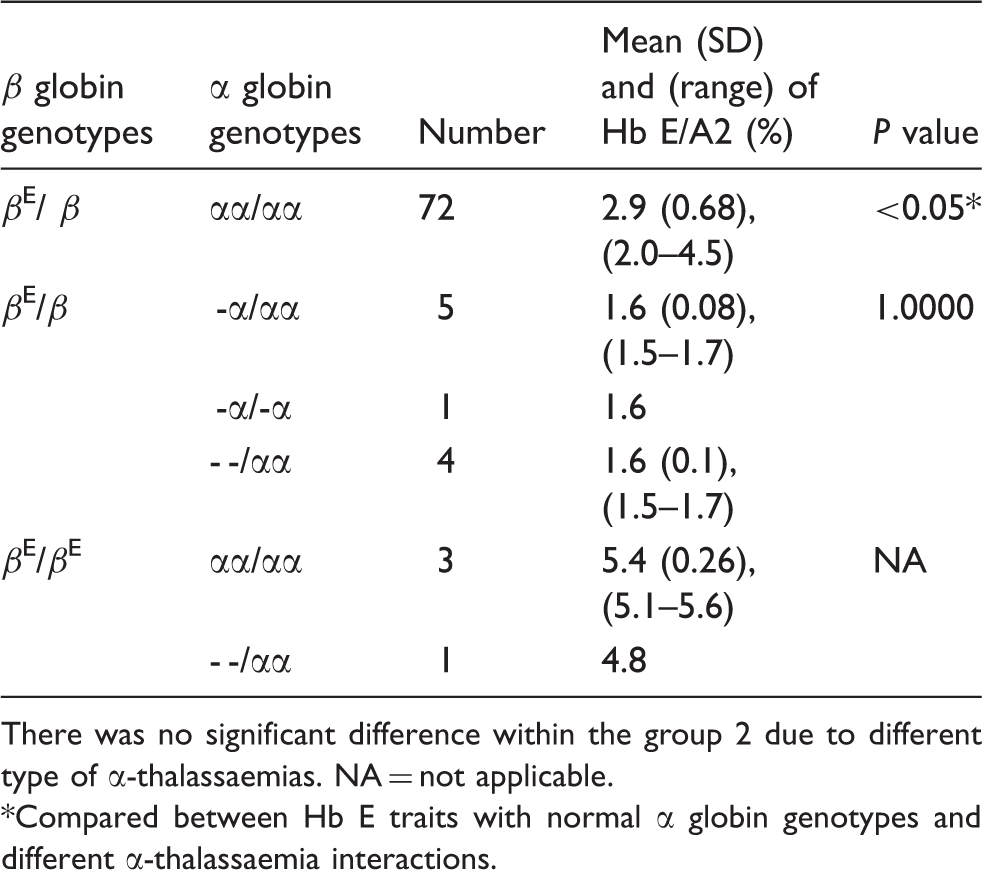
There was no significant difference within the group 2 due to different type of α-thalassaemias. NA = not applicable.
Compared between Hb E traits with normal α globin genotypes and different α-thalassaemia interactions.
Possible effect of haemoglobin E mutation to the amount of haemoglobin Bart’s detected
Effects of the β-globin gene mutations on the amount of Hb Bart’s.
Diagnostic value of isoelectric focusing for heterozygous β-thalassaemia in the newborns
Hb A was used to identify fetuses with heterozygous β-thalassaemia using IEF in immobilized pH gradients according to Manca et al. 19 Normal fetus had higher concentration of Hb A than the fetuses with heterozygous β-thalassaemia. 20 To evaluate this further in our setting, we compared percentages of Hb A between seven β-thalassaemia carriers with 131 normal individuals; all of these β thalassaemia traits were β0 mutations comprising five cases with codon 41/42 mutation (HBB:c.126_129delCTTT) and two cases with codon 17 mutation (HBB:c.52A > T). Newborns with heterozygous β-thalassaemia tend to have lower concentration of Hb A than the normal newborn (data not shown). However, there was no difference on the average of Hb A between normal and with heterozygous newborns (P = 0.10562). Therefore, there is no diagnostic value of using IEF and system described herein to detect for β thalassaemia trait at neonatal stage.
Cut-off level for diagnosis of heterozygous α+-thalassaemia
In our study, only one individual with heterozygous α+-thalassaemia was not detected with Hb Bart’s, despite Hb Bart’s was also detected at a low amount in 96 cases with normal α globin genotype. Thus, an ROC curve was constructed to examine the diagnostic concentrations of Hb Bart’s for α+-thalassaemia carriers. Newborns with normal α–globin genes served as the reference group, whereas those with α+-thalassaemia carriers were treated as the patient group. The area under the curve of the ROC curve which was 0.835 with their 95% confidence interval was evaluated as measures of diagnostic accuracy (Figure 4). The optimal cut-off level of Hb Bart’s of >0.25% (as measured by Isoscan® Imaging machine) seems to be most appropriate for newborn screening for α+-thalassaemia carriers with the sensitivity of 92.9% and specificity of 74.0%.
ROC curves for the diagnosis of heterozygous α+-thalassaemia based on Hb Bart’s concentrations (area under curve 0.835).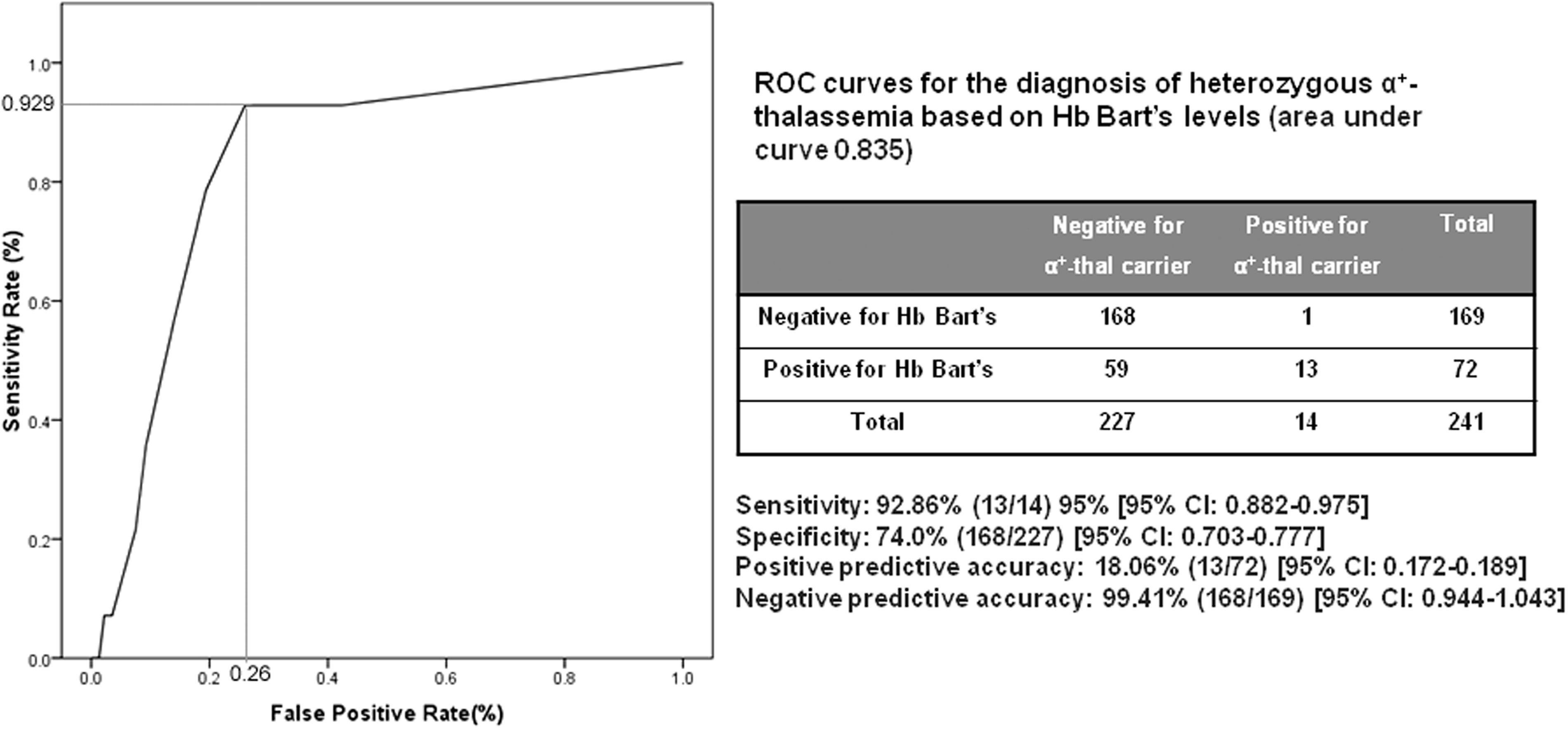
Criterion setting for thalassaemia and haemoglobinopathies diagnosis by isoelectric focusing
Standard reference range for presumptive diagnosis of α-thalassaemia traits, Hb H and Hb E disorders using the concentrations of Hb Bart’s and Hb E.

*Known as Hb AE Bart’s disease.
Known as Hb EF Bart’s disease.
Stability of haemoglobins in dried blood spot for isoelectric focusing analysis
In our routine child care setting, dry blood spots were collected at any medical care centres where delivery services are available. However the laboratory screenings are usually performed centrally at referral or regional centres with standardized facilities. Therefore, it is important that the blood sample must be in a good condition and kept well before transfer. There was an increasing detection of ageing haemoglobin bands by the IEF by day 7 onwards (Figure 5). Both Hb Bart’s and Hb E remained detectable up to 3 weeks but their concentrations changed after 7 days. Therefore, IEF screening should be performed within 1 week in order to accurately identify and quantify haemoglobins.
Stability of Hb Bart’s and Hb E and development of ageing haemoglobins on stored dry blood spots for IEF newborn screening.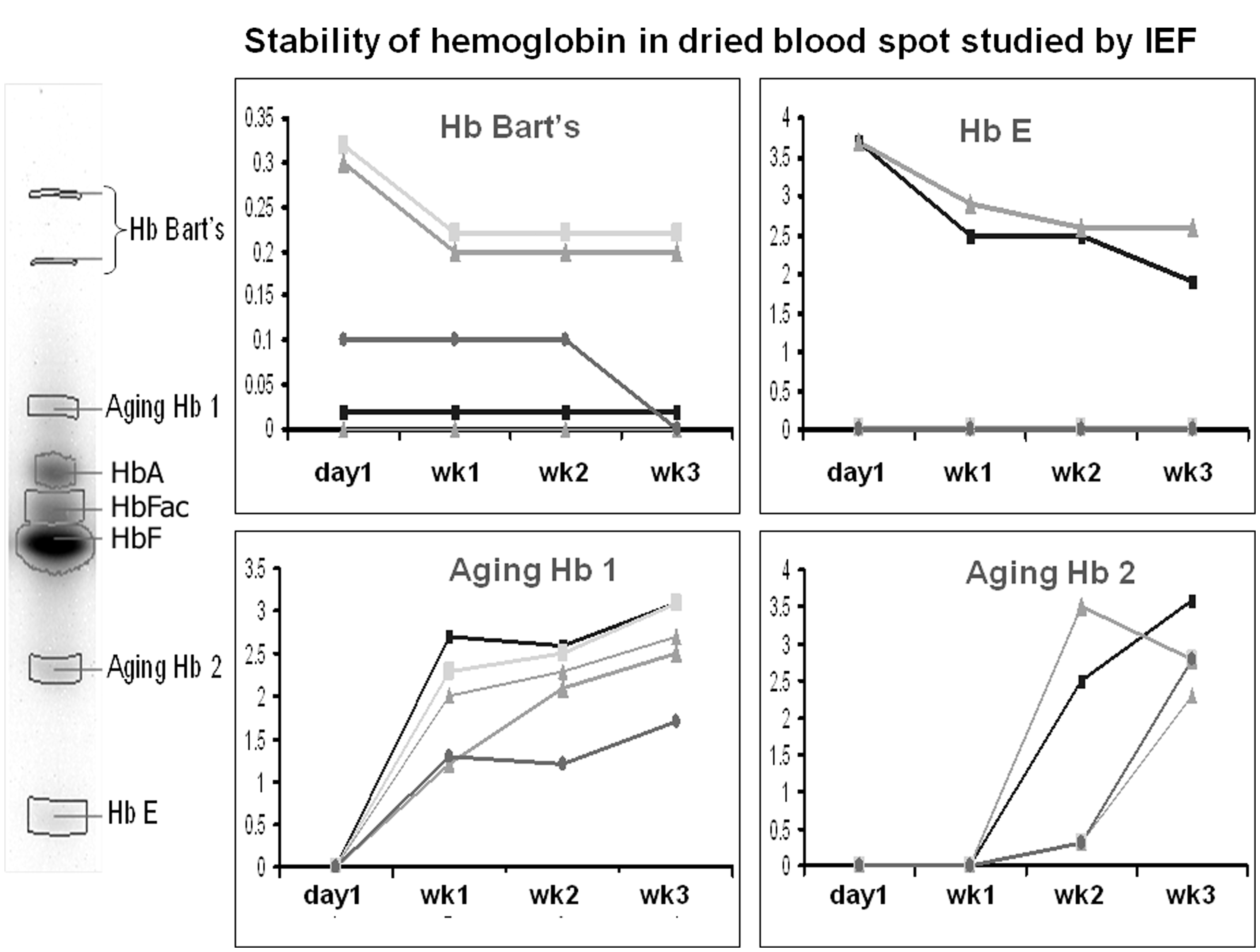
Discussion
In several developed countries, newborn screening programmes have been proven to be successful for early detection of several important haemoglobinopathies including sickle cell disorder (SCD).21–25 Moreover, using the presence of Hb Bart’s due to a tetrameric formation of the γ globin chains (γ4) have been previously shown to identify coinheritance of α-thalassaemias and Hb H disease at birth.16–18,26 In addition, it might be possible to detect Hb E and β-thalassaemia traits using the quantity of haemoglobins identified.15,19 Although SCD appeared to be rare in Thailand and Southeast Asian countries, 4 however, early identification of our common thalassaemia diseases such as Hb H, β-thalassaemia major (β0/β0) and Hb E/β thalassaemia (βE/β0) since birth can help these families to manage their child earlier and more appropriately before these patients would develop their anaemic symptoms. Moreover, early detection of carrier status with efficient and maintained national thalassaemia database linked to our health care service could be another measure to improve our screening programme in the near future.
In the past recent years, several groups in Thailand have evaluated the clinical applications of haemoglobin analysis as a newborn screening tool to identify thalassaemia carriers and diseases.27–29 However, there were several limitations associated with these studies; all reports mainly used cord blood instead of dry blood spots, which is different from current practice in our country. Due to different source of blood and associated preservative and collection process, it might be possible that the reference ranges and cut-off for each haemoglobin might be different if such criteria were applied into a routine service. In one study, high-performance liquid chromatography (HPLC) was selected as a method for haemoglobin analysis, 27 however, such a technology has several drawbacks; based on the reported system (Primus Corp, Kansas City, Mo), this is different from the system commonly used in our country (β-thalassaemia short programme kit on a BioRad Variant, Hercules, California, USA). Hb Bart’s was presumably detected and measured after a local adjustment and this may not be applicable for other laboratories outside the standard reference setting. Moreover, the cost per run for HPLC would be around 10–20 USD in Thailand. Recently, a screening programme using capillary electrophoresis (CE) has been proposed. Again, the cost per test (12 USD) remains a major obstacle to a nationwide programme. 29 At present, IEF is probably the method of choice in most clinical laboratories for detecting haemoglobinopathies because it is inexpensive (1.5 USD per one test), easy-to-use and can distinguish a healthy subject from a possibly afflicted individual needing further testing to diagnose a possible haemoglobinopathy. 23 Although a comprehensive molecular testing used in this study can provide a definitive diagnosis, however it costs approximately 150 USD per sample and this could limit its application in a routine service.
In this study we further applied the Isoscan® Imaging System to quantify the amount of haemoglobins to presumably diagnose for at least 10 different genotypes based on our recommended criteria (Table 4). Our proposed diagnostic scheme was validated and compared with the definite genotype data derived from our comprehensive molecular analyses in 350 newborns. The findings provided a clear cut reference range for each genotype category and most of the data were consistent with previous studies.17,18,27,28 The amount of Hb Bart’s detected, rather than the presence, is more important to accurately diagnose α-thalassaemia. 27 However, diagnosis of deletional α+-thalassaemia carrier (-α/αα) by Hb Bart’s at birth remains problematic; one of our 14 α+-thalassaemia traits (7.14%) had undetectable Hb Bart’s in this study while all three Hb CS carriers had Hb Bart’s ≥1%. Consistently with previous reports by Tanphaichitr et al. 17 and Lie-Injo et al. 16 that 12.7% and 6.25% of their α+-thalassaemia traits, respectively, had unidentifiable Hb Bart’s by starch gel and cellulose acetate electrophoresis. Using ROC curve analysis, we suggested to use a cut-off >0.25% Hb Bart’s to presumptively diagnosed α+-thalassaemia trait with a sensitivity of 92.86%. This should be practically, although not perfectly, applicable for a routine laboratory setting. However, a definitive diagnosis by DNA study in cases with positive screening is still important due to a rather low specificity of this cut-off (74%).
Diagnosis of Hb E disorders including heterozygous and homozygous Hb E with or without α-thalassaemia interactions is made possible using our models. We provided further evidence that a limitation of α-globin chain production could result in a decrease in Hb E detection from neonatal period. Our data further elaborate an observation in a recent study by Charoenkwan et al., 28 using a similar approach but different source of blood samples with a better precision and no overlapping range. In the previous study, 28 the concentrations in Hb E traits with ‘so-called normal’ α globin genotype was somewhat overlapping with those of single or two α gene missing (0.9–2.9% vs. 0–0.5% and 0.5–0.8%, respectively). It is plausible that the molecular analysis performed in the previous study was not comprehensive enough compared to our study, therefore some cases with ‘genuine’ α thalassaemia defects might have been missed as ‘normal’ α globin genotype. In addition, some individuals identified with single α globin gene defect might be in fact carried another undetected α thalassaemia alleles on another chromosome due to the study’s limitation.
In conclusion, this study provides a standard reference range to identify thalassaemia diseases and simple and double thalassaemia carriers for newborn screening in Thailand with a diagnostic coverage of 97% of Thai population. Moreover, our model described herein can be easily adopted into our current national programme on neonatal screening since our reference range was based on dry blood spots. We also recommend keeping the filter papers at −20℃ and performing the IEF within 7 days after sample collection. This study could further improve our management and standard of dedicated patients care on thalassaemia diseases from early childhood, increase efficacy and furnish a long-term success of thalassaemia and haemoglobinopathies screening, prevention and control policy, and control of new birth with severe thalassaemia syndrome at a nationwide level. This ‘Siriraj-Newborn Screening Strategy’ for thalassaemia could be applied to other developing countries in Southeast Asia where the prevalence of globin gene disorders are highly apparent and health resource is limited.
Limitation of the study
Our study was limited since there was no individual with Hb E/β thalassaemia identified; however, based on our data, we would predict such individual, without α-thalassaemia interaction, to have the profile of Hb F and E with the concentrations of Hb E 2–4.5%. In addition, we failed to clearly diagnose individuals with β-thalassaemia carriers. Even by using a recent recommended Hb A cut-off at <15% of total haemoglobins, 20 42% of our β-thalassaemia carriers had the concentrations of Hb A above this guideline. Moreover, in our study, no data on gestational age of the mothers were available and this factor plays an important role on detection of Hb A at neonatal period. 20 Further study is warranted to improve the quality of IEF gel to determine more accurately the concentrations of total haemoglobin against Hb A.
Footnotes
Acknowledgements
We thank Worrawut Chinchang for his excellent technical expertise on developing molecular tests used in this study.
Declaration of conflicting interests
None.
Funding
This research was supported by a Siriraj Grant for Research Development and Medical Education, the Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, National Research University grant, BIOTEC, Thailand and the Thailand Research Fund (VV). PP is supported a grant from Department of Clinical Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University and Phramongkutklao Hospital.
Ethical approval
The ethics committee of the Ramathibodi and Phramongkutklao Hospitals approved this study (No.2008/1051).
Guarantor
VV.
Contributorship
VV designed the study, analysed the data, wrote the manuscript and was responsible for all figures and tables. PJ was involved in protocol development, gaining ethical approval, performed haemoglobin and molecular analyses, organized and analysed clinical and molecular data and prepared the first draft of the manuscript. WG and SR performed molecular analysis. YS and SC collected blood samples and informed consent. KT was involved in protocol development and provided helpful comments and mentoring. All authors approved the final version of the manuscript before submission.