
Abstract
The porphyrias are a group of mainly inherited metabolic conditions that result from partial deficiency of individual enzymes in the haem biosynthesis pathway. Clinical presentation is either with acute neurovisceral attacks, skin photosensitivity or both, and is due to overproduction of pathway intermediates. The primary diagnosis in the proband is based on biochemical testing of appropriate samples, preferably during or soon after onset of symptoms. The role of genetic testing in the autosomal dominant acute porphyrias (acute intermittent porphyria, hereditary coproporphyria and variegate porphyria) is to identify presymptomatic carriers of the family specific pathogenic mutation so that they can be counselled on how to minimize their risk of suffering an acute attack. At present the additional genetic factors that influence penetrance are not known, and all patients are treated as equally at risk. Genetic testing in the erythropoietic porphyrias (erythropoietic protoporphyria, congenital erythropoietic porphyria and X-linked dominant protoporphyria) is focused on predictive and preconceptual counselling, prenatal testing and genotype–phenotype correlation. Recent advances in analytical technology have resulted in increased sensitivity of mutation detection with success rates of greater than 90% for most of the genes. The ethical and consent issues are discussed. Current research into genetic factors that affect penetrance is likely to lead to a more refined approach to counselling for presymptomatic gene carriers.
Introduction
The porphyrias
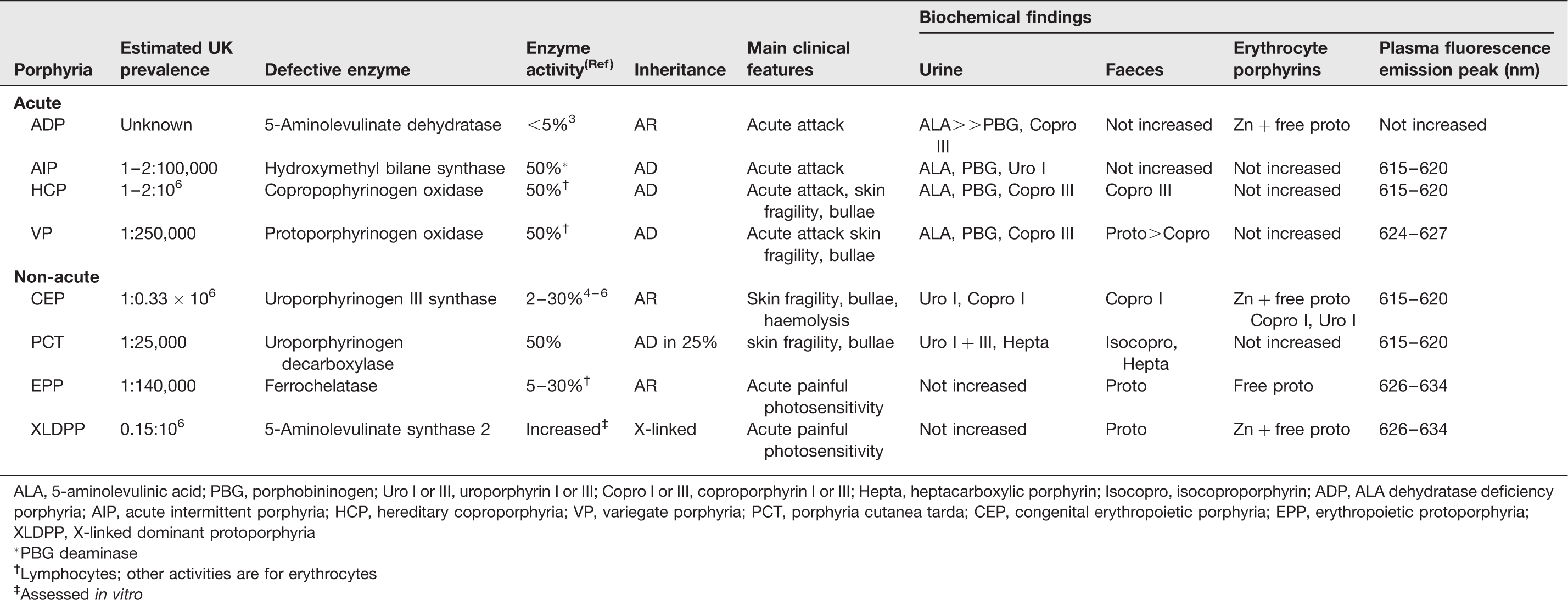
ALA, 5-aminolevulinic acid; PBG, porphobininogen; Uro I or III, uroporphyrin I or III; Copro I or III, coproporphyrin I or III; Hepta, heptacarboxylic porphyrin; Isocopro, isocoproporphyrin; ADP, ALA dehydratase deficiency porphyria; AIP, acute intermittent porphyria; HCP, hereditary coproporphyria; VP, variegate porphyria; PCT, porphyria cutanea tarda; CEP, congenital erythropoietic porphyria; EPP, erythropoietic protoporphyria; XLDPP, X-linked dominant protoporphyria
*PBG deaminase
†Lymphocytes; other activities are for erythrocytes
‡Assessed in vitro
Each porphyria results from overproduction of haem precursors secondary to partial deficiency or, in X-linked dominant protoporphyria (XLDPP), increased activity of one of the enzymes of haem biosynthesis (Figure 1; Table 1). These result in specific patterns of accumulation and excretion of haem precursors that define each disorder (Table 1). Diagnosis is normally straightforward, and requires only biochemical investigation, provided the appropriate analytes are measured in the appropriate samples while symptoms are present, or soon after.7,8 Indeed, since symptoms may be non-specific, porphyria cannot be established as their cause unless haem precursor concentrations are shown to be increased. Genetic testing is rarely required to make a diagnosis of porphyria and, by itself, may be misleading if a mutation is not found or unclassified variants are identified. Genetic analysis does not identify mutations in all unequivocally diagnosed cases and therefore cannot be used to exclude a diagnosis of porphyria. In addition, low clinical penetrance in the autosomal dominant porphyrias means that identification of a mutation does not necessarily indicate active porphyria. However, DNA analysis is now the method of choice for presymptomatic diagnosis, family studies and for predictive counselling. This review summarizes current knowledge of the molecular genetics of the porphyrias and describes the use of genetic testing in these disorders.
Haem biosynthetic pathway. Enzymes are shown in solid fill, acute porphyrias (striped background) ADP, 5-aminolevulinic acid dehydratase deficiency porphyria; AIP acute intermittent porphyria; HCP, hereditary coproporphyria; VP, variegate porphyria. Cutaneous porphyrias (circles background): XLDPP, X-linked dominant protoporphyria; CEP, congenital erythropoietic porphyia; PCT, porphyria cutanea tarda; EPP, erythropoietic protoporphyria. Clear background: XLSA, X-linked sideroblastic anaemia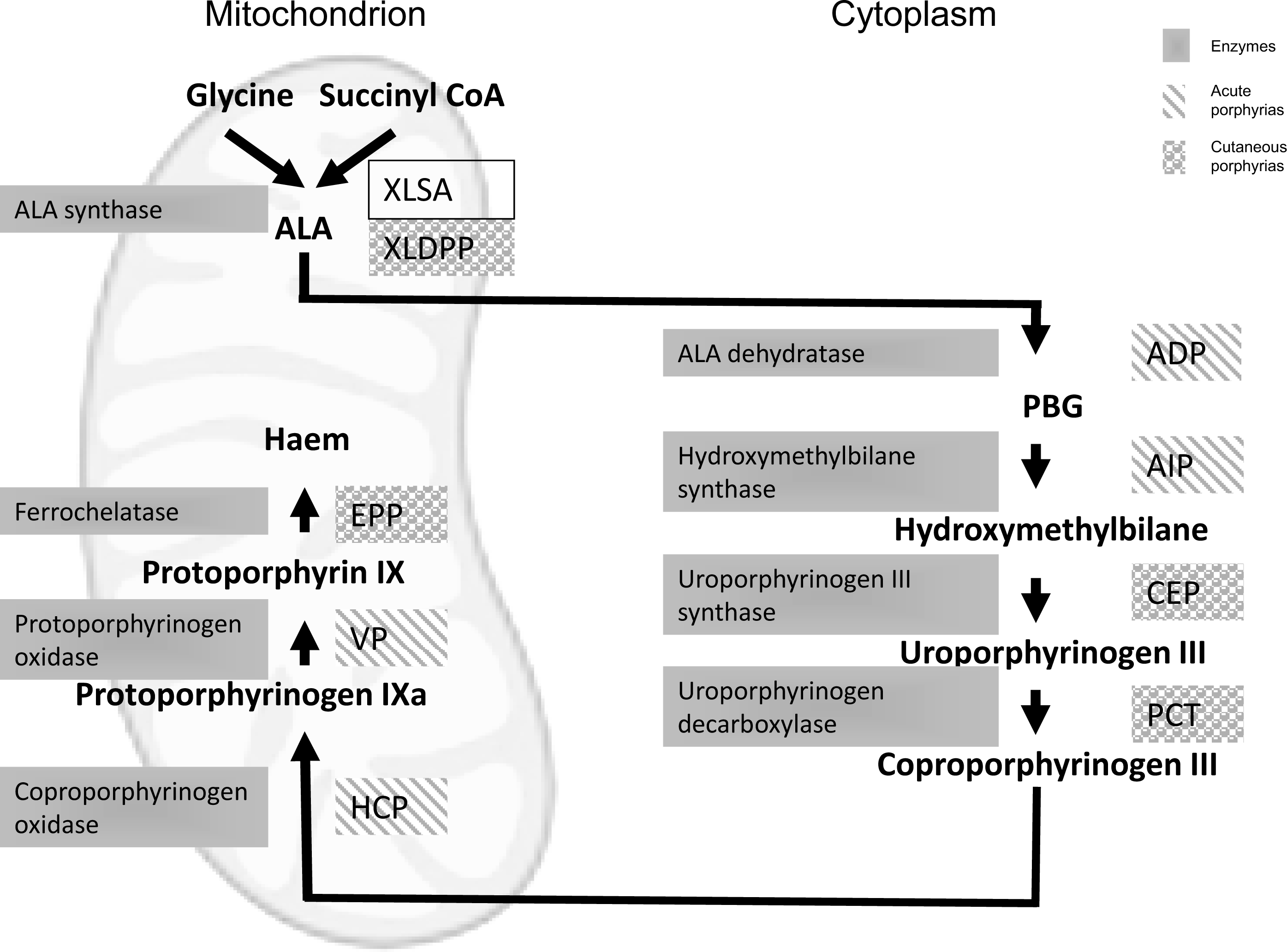
Gene structure and expression
Human genes encoding enzymes of haem biosynthesis
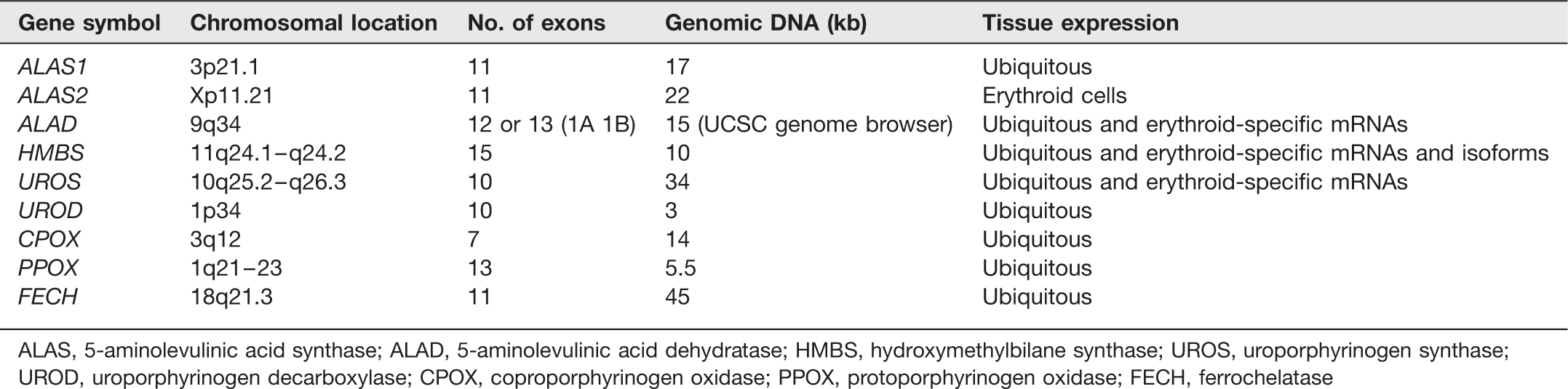
ALAS, 5-aminolevulinic acid synthase; ALAD, 5-aminolevulinic acid dehydratase; HMBS, hydroxymethylbilane synthase; UROS, uroporphyrinogen synthase; UROD, uroporphyrinogen decarboxylase; CPOX, coproporphyrinogen oxidase; PPOX, protoporphyrinogen oxidase; FECH, ferrochelatase
ALAS has both erythroid and non-erythroid genes17,18 with the next three genes of the haem biosynthetic pathway (ALAD, HMBS, UROS) having dual promoters18–20 (Figure 2). This enables the genes in the liver and the erythroid cells to be regulated according to the organs differing haem requirements. Most haem synthesis takes place in the developing red cells in the bone marrow with about 15% produced in the liver for the formation of haem-containing enzymes.
21
In the liver, most haem biosynthetic enzymes are turned over rapidly enabling the liver to respond effectively to changing metabolic requirements. ALAS1 is the rate-limiting enzyme in the production of hepatic haem
17
and is controlled by negative feedback regulation by the intracellular haem pool. In erythroid cells the rate of ALAS2 synthesis is regulated to permit a high level of haem synthesis and is linked to the availability of iron
22
and is not inhibited by haem.
23
5-Aminolevulinic acid dehydratase (ALAD), hydroxymethylbilane synthase (HMBS) and uroporphyrinogen synthase (UROS) genes are transcribed from different promoters to produce ubiquitous and erythroid isoforms of the enzymes in these different tissues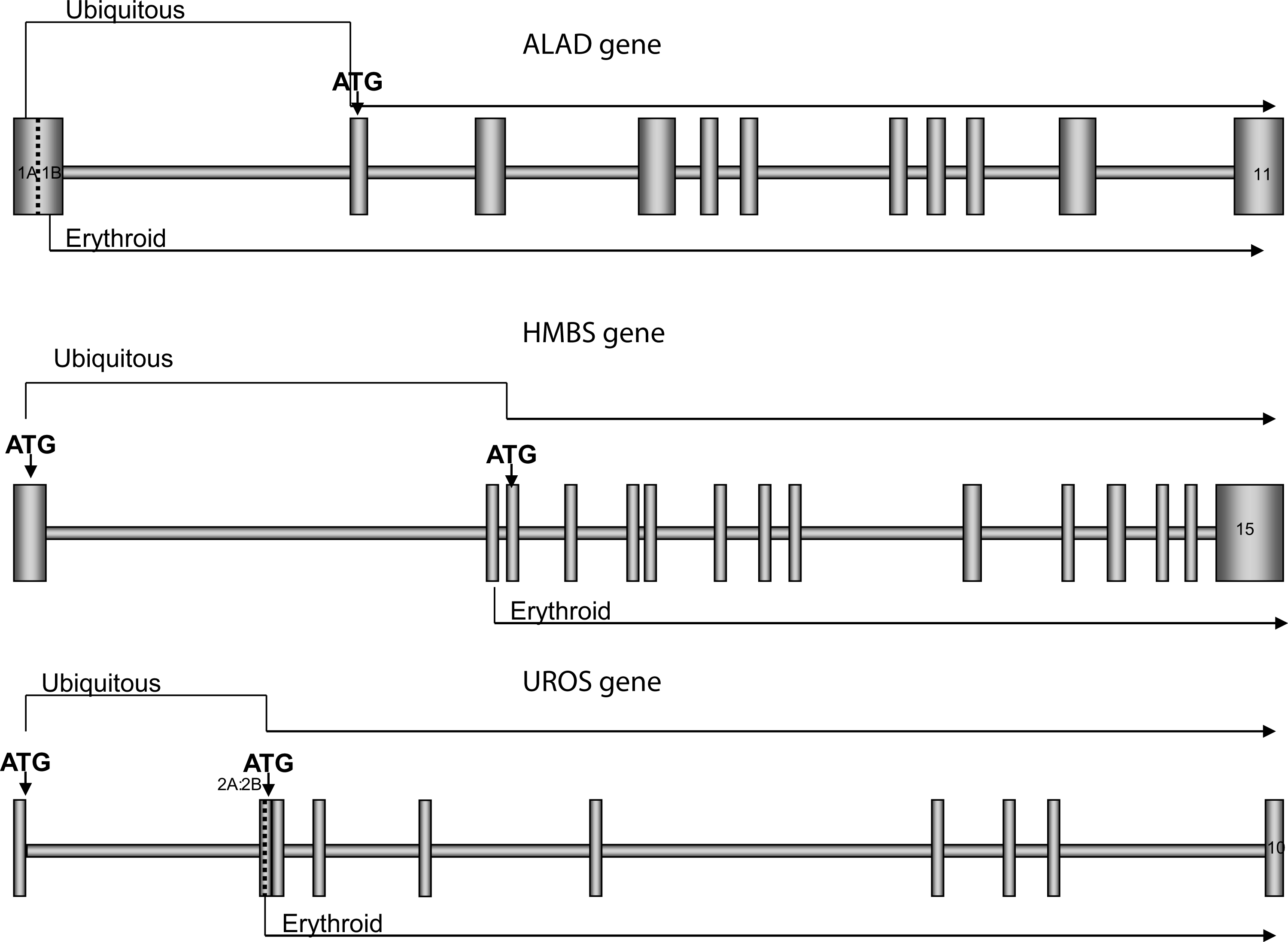
Autosomal dominant porphyrias
Four porphyrias are inherited in an autosomal dominant pattern: acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP) and familial porphyria cutanea tarda (F-PCT). At the molecular level, all show extensive allelic heterogeneity with most mutations being restricted to one or a few families although founder mutations have been identified in some countries, notably those that underlie the high prevalence of AIP in Sweden, 24 VP in South Africa 25 and F-PCT in Norway. 26 Enzyme activities are reduced to around 50% of normal, indicating near or complete haplodeficiency.27,28 All four disorders show low clinical penetrance indicating that environmental factors and, probably, genes at other loci are important in determining their presentation. Family studies in France 1 and the UK suggest that about 10–20% of affected individuals develop symptoms but figures as high as 50% have been reported for AIP from Sweden 29 and, when minor skin lesions are taken into account, 40% for VP from South Africa.30,31 As might be predicted for a heterogeneous haplodeficient disorder, no clear genotype–phenotype correlation has yet been established in any autosomal dominant porphyria though there are reports that some mutations may be associated with high penetrance.29,31
There is evidence that mutations for all four disorders are more common in western European populations than the prevalence of the diseases would suggest. In France, one in 1675 blood donors carries an HMBS gene mutation. 27 Consistent with this high gene frequency, rare homozygous or compound heterozygous forms, so-called ‘homozygous’ variants, have been described for each autosomal dominant porphyria.32–36 Also consistent with a high prevalence of asymptomatic heterozygotes in the population are the high frequency of sporadic presentation, with less than 50% of patients having a family history of overt porphyria, unexplained by de novo mutation, and the rare occurrence of co-inheritance of two porphyrias, so-called dual porphyria.37,38
Autosomal dominant acute porphyrias
General features
AIP, HCP and VP are characterized by the episodic occurrence of life-threatening acute neurovisceral attacks (Table 1) which are identical in all three conditions. In VP and HCP, bullous skin lesions are present during an acute attack in 10–50% of patients or may be the only clinical manifestation (Table 1).
1
This is rare in HCP but, in the UK, about 60% of patients with VP present only with skin lesions, identical to those of PCT.
39
Acute attacks are rare before puberty and often provoked by identifiable precipitants, notably certain drugs, endocrine factors and alcohol. Their management and prevention has recently been reviewed.
1
Preventing acute attacks by advising patients to avoid porphyrogenic drugs [European Porphyria Network:
Molecular genetics
More than 342 mutations have been identified in the HMBS gene in AIP, about 52 in the CPOX gene in HCP and more than 150 in the PPOX gene in VP (Human Gene Mutation Database:
Indications for molecular investigation
Sensitivity of mutation detection
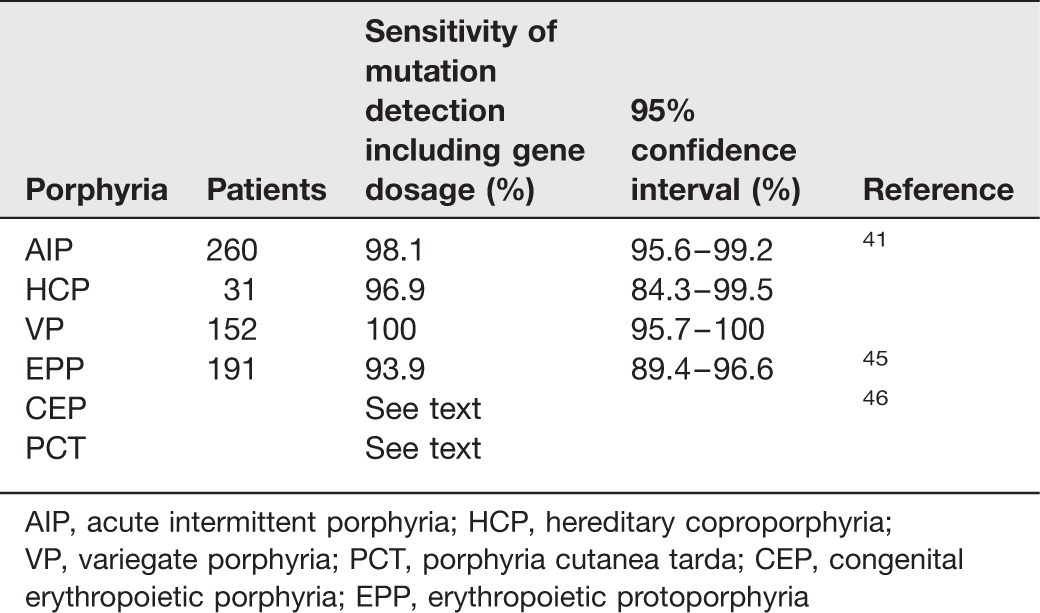
AIP, acute intermittent porphyria; HCP, hereditary coproporphyria; VP, variegate porphyria; PCT, porphyria cutanea tarda; CEP, congenital erythropoietic porphyria; EPP, erythropoietic protoporphyria
Family studies: Presymptomatic diagnosis of affected relatives is an essential part of the management of families with AIP, HCP or VP. Provided the family mutation can be identified, DNA analysis is now the method of choice, having 100% sensitivity for this purpose.1,47 It has greater diagnostic accuracy than erythrocyte HMBS activity assay for AIP, plasma fluorescence scanning for VP and faecal porphyrin analysis for HCP and, unlike metabolite assays, is applicable before puberty. 48 In contrast to biochemical investigations, it can exclude inheritance of the family mutation with a high degree of certainty but, since the rest of the gene is not sequenced there is a theoretical possibility of the patient having a different mutation within the same gene. Biochemical investigation can be used to decrease the number of adult relatives that require DNA analysis, particularly in VP where plasma fluorescence scanning has been reported to identify 76% of asymptomatic gene carriers. 49 Faecal coproporphyrin isomer analysis may similarly reduce the need for mutation testing in adult relatives of HCP patients. 50
Retrospective investigation of suspected acute porphyria: Occasionally, patients who are currently asymptomatic may require investigation to confirm or exclude a past history of acute porphyria in themselves or in a relative who is no longer available for investigation. Because symptoms may be followed by full biochemical remission, normal metabolite measurements do not exclude the diagnosis, particularly in AIP, and enzyme measurements lack sufficient diagnostic accuracy. 51
In this situation, DNA analysis may identify a disease-specific mutation or, for mutation-negative patients, in combination with biochemical investigation, allow the patient's risk of being affected to be assessed. 41 Our current practice does not include analysis of all three genes as this is not considered cost-effective.
Homozygous acute porphyrias
The number of gene carriers for all three autosomal dominant acute porphyrias is sufficiently high for patients to inherit either two acute porphyrias or mutations for the same disease on each allele. The latter patients are either homozygotes or compound heterozygotes and at least one of the mutations must be associated with some residual enzyme activity in order to sustain life. Homozygous AIP is usually associated with severe, progressive central and peripheral neurological deterioration after birth. 33 Homozygous VP presents with bullous skin lesions in childhood, often associated with skeletal and neurological defects.52,53 Homozygous HCP may present with acute neurovisceral attacks in childhood 54 while a variant, harderoporphyria, causes neonatal haemolytic anaemia and mild photosensitivity. 55 Patients are either heteroallelic or homoallelic for a missense mutation, p.Lys404Glu, in exon 6 of the CPOX gene that impairs the sequential decarboxylation of coproproporphyrinogen III resulting in increased faecal excretion of the tricarboxylic intermediate, harderoporphyrin. Molecular genetic analysis is essential to confirm the diagnosis in these rare conditions.
Porphyria cutanea tarda
General features
PCT is by far the commonest porphyria with over 100 new cases per year in the UK. Patients usually present with skin fragility and bullae on sun-exposed skin. 56 Evidence of liver dysfunction and some degree of iron overload is common. Associated risk factors include high alcohol intake, hepatitis C infection, haemachromatosis and use of oestrogens.57–60 Treatment is by depletion of iron stores, usually by repeated phlebotomy, or with low-dose oral chloroquine. Both are equally effective in all patients and produce prolonged remission or cure.60–62 The disease results from reversible inactivation of uroporphyrinogen decarboxlase (UROD) in the liver; symptoms do not occur until activities are below 50% of normal. Inactivation is reported to be due to an inhibitor formed by iron-dependent oxidation of uroporphyrinogen. 63
Molecular genetics
Enzymatic and molecular investigations have revealed the existence of two main types of PCT. 56 Most patients have the sporadic form (S-PCT) in which UROD deficiency is restricted to the liver and the UROD gene is normal. About 25% of patients have the autosomal dominant form, familial PCT (F-PCT) in which UROD activity is decreased in all tissues. Over 108 disease-specific mutations have been identified in the UROD gene in F-PCT and its rare ‘homozygous’ variant, hepatoerythropoietic porphyria (HEP).64,65 Mutations in F-PCT decrease UROD activity by 50%. An additional decrease in hepatic UROD is required to produce symptoms which is brought about by the inactivation mechanism described above which is common to both types of PCT. The need for additional inactivation and the frequent presence of the same risk factors in both types of PCT explains, at least in part, the low clinical penetrance of F-PCT. The sensitivity of mutation identification in F-PCT probably exceeds 95% but is difficult to determine because there is no method for unequivocally distinguishing F-PCT from S-PCT which may itself rarely cluster in families. 66
Indications for molecular investigation
Molecular analysis of the haemochromatosis gene should be part of the initial investigation of all patients with PCT in the UK where about 20% are homozygous for the p.Cys282Tyr (C282Y) mutation.59,60 Homozygotes should be treated by phlebotomy, not with chloroquine, and subsequently monitored for re-accumulation of iron.
Mutational analysis of UROD is not needed for the diagnosis of PCT except in the rare instances when HEP is suspected. 65 There is currently debate about whether all patients with PCT should be offered testing to identify those with F-PCT.1,26,67 Mutational analysis of UROD has greater diagnostic accuracy than measurement of erythrocyte UROD activity for this purpose.26,68 Differentiation of F-PCT from S-PCT has little benefit for the individual patient since the response to treatment and prognosis of the two forms is similar. 67 However, subsequent screening of their families would allow asymptomatic affected relatives to be counselled about the need to avoid risk factors. As yet, there is no published evidence for the benefits of managing families in this way. Some patients and relatives may wish to know whether their disease is inherited. 26 In these, DNA analysis of UROD in the proband may be justified after appropriate genetic counselling. Current practice in the UK is to undertake only biochemical analyses except in exceptional circumstances.
Autosomal recessive porphyrias
Two rare porphyrias, ADP 69 and congenital erythropoietic porphyria (CEP) are inherited in an autosomal recessive pattern and a third, erythropoietic protoporphyria (EPP), is a recessive disorder which due to a prevalent hypomorphic allele behaves in a pseudodominant fashion. 70 XLDPP, although an X-linked disorder, is included in this section as it is convenient to consider it as a form of EPP.
Enzyme activities in the autosomal recessive porphyrias are decreased in all tissues to around 30% of normal or less. In contrast to the dominant porphyrias, all three disorders are fully penetrant with only very rare exceptions 71 and, in common with other autosomal recessive disorders, some genotype–phenotype correlation is apparent.
ALA dehydratase deficiency porphyria
ADP is a very rare acute porphyria; only six families have been reported worldwide. 69 Patients are usually compound heterozygotes for ALAD mutations. Up to 2% of the population may be heterozygotes, who although asymptomatic, may be more susceptible to toxicity from lead and other chemicals that inhibit this enzyme. 72
Congenital erythropoietic porphyria
General features
CEP is the most severe of the cutaneous porphyrias, affecting about three per 10 million of the UK population. It usually presents with red urine, severe blistering and haemolytic anaemia in infancy. Patients may develop photo-mutilation and become transfusion-dependent. As with all recessive conditions, the phenotype can vary depending on residual enzyme function and more severe forms presenting in utero with hydrops foetalis as well as mild, later onset forms clinically resembling PCT are described.73–75 Treatment depends on photo-protection and other supportive measures.76,77 Allogeneic bone marrow transplantation is curative but is usually reserved for the more severe patients with haemolytic anaemia and should be undertaken at a young age wherever possible.
Molecular genetics
Affected individuals are homozygous or compound heterozygous for UROS mutations of which about 45 have been reported (HGMD:
Indications for molecular investigation
Mutation analysis of the UROS gene, although not required for diagnosis, may be useful for assessing prognosis, particularly in helping to decide whether allogeneic bone marrow transplantation is indicated. Family members who are heterozygotes for CEP mutations are asymptomatic and have successfully acted as donors. 79 Prenatal diagnosis in families who already have an affected family member can be helpful, but is dependent on identification of both mutations. Where these are not identified, measurement of uroporphyrin I in amniotic fluid may be informative. 80
Erythropoietic protoporphyria
General features
EPP is a disorder in which accumulation of protoporphyrin in erythrocytes, skin, liver and other tissues leads to lifelong, acute painful photosensitivity and, in about 2% of patients, severe liver disease. Symptoms usually start in early childhood. Chronic skin lesions are minor and skin fragility is absent.81,82 The most effective treatment is the prevention of the photosensitive reaction by avoiding sunlight, skin protection with clothing and sunscreen ointments and by increasing skin pigmentation.83,84 Severe liver disease usually requires transplantation.85,86
Molecular genetics
Excess accumulation of free protoporphyrin results from partial deficiency of ferrochelatase (FECH) activity. In the UK, most EPP patients are compound heterozygotes for a hypomorphic IVS3-48C allele that produces a truncated unstable mRNA, reducing activity by 20–30%,
87
and a deleterious FECH mutation (Figures 3a and b) that abolishes or markedly decreases enzyme activity.
45
This combination reduces overall FECH activity below a threshold of 35% of normal at which symptoms occur.
88
The population frequency of the IVS3-48C allele varies, ranging from less than 1% in West Africa to 45% in Japan,
88
and correlates with the prevalence of EPP and the frequency of parent to offspring transmission. In the UK, where 13% of the population carry the low expression allele,
45
30–35% of unrelated patients have an affected relative and EPP is present in more than one generation in about 5% of families (Figure 3c).
81
The inheritance of EPP. (a) Family with an affected child who has inherited a mutation (M) from the mother and a low expression allele (C) from the father. Both parents are asymptomatic. (b) Affected mother but no children have inherited overt EPP. (c) Affected mother, father has a low expression allele (C). The risk of having a child with overt EPP is one in four. (d) Mother and father both have mutations in the FECH gene (M). The risk of having a child with overt EPP is one in four. Both parents are asymptomatic. N denotes ‘normal’ FECH allele. T denotes thymine at position IVS3-48, C denotes cytosine at position IVS3-48 (the low expression polymorphism). Shaded box/circle denotes symptomatic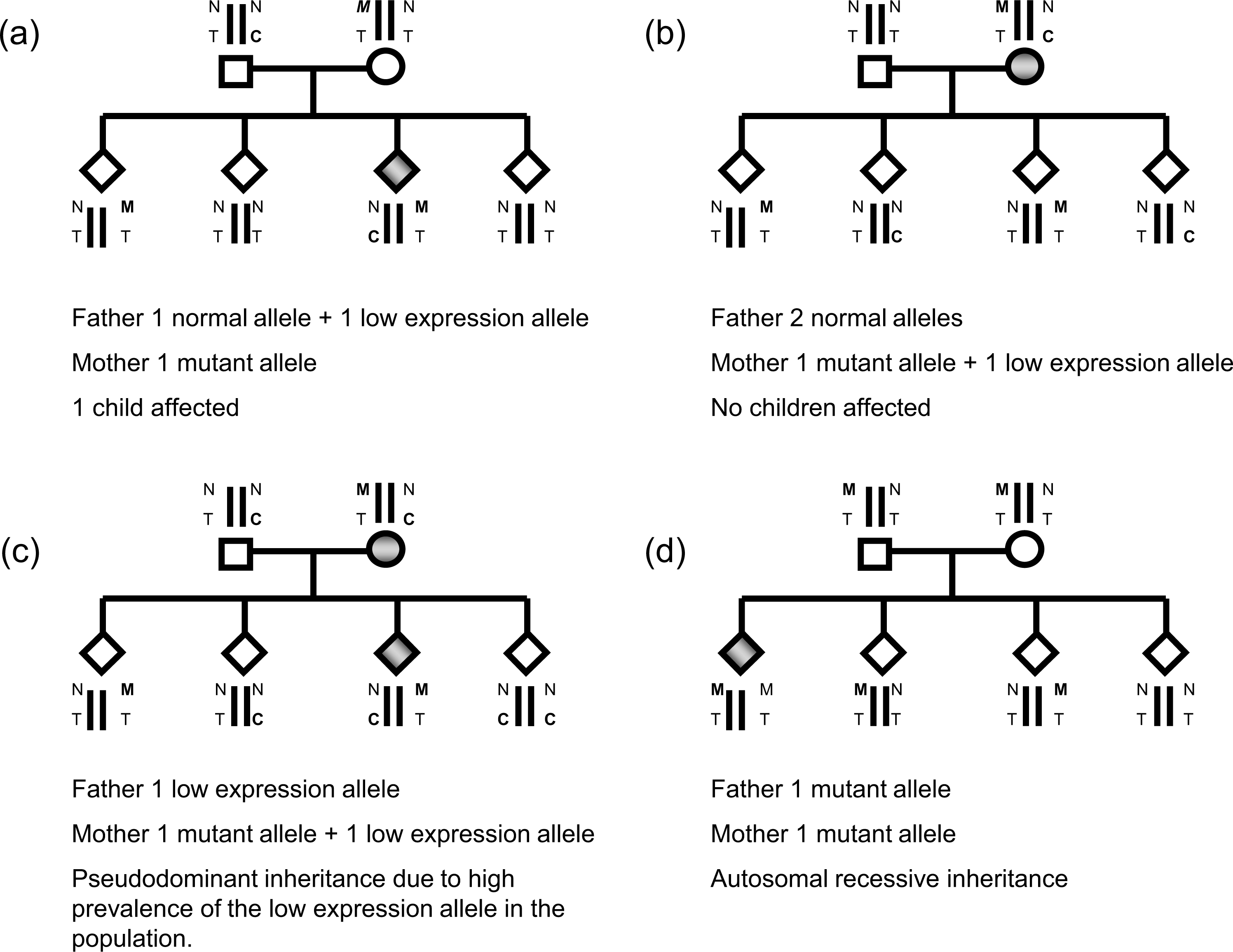
Patients from about 4% of families are homozygous or compound heterozygous for deleterious mutations (Figure 3d) and characteristically have residual FECH activities of around 10% of normal. The frequency of the IVS3-48C allele in this group of patients does not differ significantly from the general population. 45
More than 130 FECH mutations have been identified in EPP (Human Gene Mutation Database:
In 2% of UK families, FECH activity is normal and a gain of function mutation in ALAS2, the gene encoding the rate-limiting enzyme of erythroid haem synthesis, results in increased flux through the pathway. The resultant accumulation of free and zinc-chelated protoporphyrin, which may be due to reduced availability of iron and or zinc within the mitochondrion, gives rise to a distinct protoporphyria, named X-linked dominant protoporphyria (XLDPP). 89 Interestingly, loss of function ALAS2 mutations cause X linked sideroblastic anaemia (XLSA). 90
Genotype:phenotype correlations
A molecular explanation has been sought for two unusual phenotypes of EPP: severe liver disease and palmar keratoderma. The risk of liver disease is increased in the rare patients who are homozygous or compound heterozygous for deleterious mutations, even though they are clinically indistinguishable from the more common form of EPP with regard to severity of photosensitivity. Five of 21 (24%) reported patients with this molecular type of EPP had severe liver disease. However, the majority of patients with severe liver disease come from the much larger group of patients with one IVS3-48C allele. There is evidence to suggest that missense mutations that retain some residual activity carry a lower risk of liver disease than mutations that abolish FECH activity.91,92 However, mutations associated with liver disease are found more frequently in patients without liver disease. Mutational analysis currently has no role in predicting the risk of liver disease in an individual patient except when it leads to identification of deleterious mutations on both FECH alleles or an ALAS2 mutation (see below). To date, all EPP patients with palmar keratoderma have been homozygote/compound heterozygotes for deleterious FECH mutations, although interestingly they had lower total erythrocyte porphyrin concentrations than patients with a single mutation and a low expression allele and none had protoporphyric liver disease.93,94 XLDPP, which is clinically indistinguishable from EPP, has a higher risk of severe liver disease than the common form of EPP in those patients so far identified. 89
Indications for molecular investigation
Patients with EPP: Mutational analysis of FECH is not required to establish the diagnosis of FECH-deficient EPP, except for presymptomatic diagnosis in infants from affected families and in the rare patients, for example, those with keratoderma, in whom erythrocyte protoporphyrin concentrations are only marginally increased. 93 However, there are arguments for including it in the investigation of all new patients, mainly to identify patients with two deleterious mutations who are at increased risk of liver disease but also for subsequent genetic counselling (see below). An additional benefit is the collection of clinical and genetic information from sufficient numbers of patients for detection of genotype–phenotype correlations that may be clinically useful in the future. The sensitivity of detection of FECH mutations in EPP is 94% (Table 3). 45 In mutation-negative patients, decreased lymphocyte FECH activity and the presence of the IVS3-48C allele infer the presence of a FECH mutation undetectable by current methods.
Mutational analysis of ALAS2 is essential to confirm the diagnosis of XLDPP and is recommended for all EPP patients where zinc protoporphyrin comprises 10% or more of the total erythrocyte porphyrin. In addition, there are very rare patients with acute photosensitivity, and increased erythrocyte free and zinc-protoporphyrin but no detectable ALAS2 mutations; the cause of their disease has yet to be determined. 45
Family studies: Screening all asymptomatic relatives for FECH mutations is not needed for the management of all families with EPP. However, mutation analysis is required for an asymptomatic adult relative who requests predictive counselling and requires prior genetic testing of the proband. This has become increasingly important given increased awareness of the pattern of inheritance of EPP and the relatively high frequency of the low expression polymorphism in the UK population.
In XLDPP, all males and most female heterozygotes have photosensitivity. Family studies by testing for ALAS2 mutations are required to identify the few asymptomatic female carriers.
Predictive counselling: It is common for patients with EPP and their asymptomatic relatives to ask for an assessment of the risk that their children will inherit EPP. For families with a child with EPP, the risk of a subsequent child having the disease is likely to be one in four, since the unaffected parent is much more likely to be heterozygous than homozygous for the IVS3-48C allele; DNA analysis is not normally required. The risk that the first child of a parent, who either has EPP or is an asymptomatic carrier of a FECH mutation, will inherit the disease depends mainly on whether the unaffected partner carries the IVS3-48C allele (Figures 3a and b). 95 If he or she is hetero-allelic for the hypomorphic IVS3-48C allele, the risk is one in four; increasing to one in two if the unaffected partner is homozygous. If this allele is absent the risk relates to the carrier frequency of FECH mutations in the general population, which although not formally assessed, is likely to be less than 1:1000. In addition, there may be other rare, as yet unidentified, hypomorphic variants. Their existence is suggested by the finding that about 1% of EPP patients with one FECH mutation are IVS3-48T homozygotes. 45
Acquired somatic mutations
A small number of patients have been described in whom either EPP or CEP has developed in association with myelodysplasia or myeloproliferative disorder.96,97 The myeloid disorder usually precedes the onset of porphyria, patients are aged 40 y or more and there is no family history of porphyria. Most of the EPP patients have an acquired full or partial deletion of chromosome 18 which results in loss of heterozygosity for FECH in a clone of haematopoietic cells.96,98 The association of CEP with myelodysplasia remains unexplained but may result from a similar mechanism.97,99
Prenatal diagnosis in the porphyrias
Prenatal diagnosis is rarely indicated in the porphyrias. In most, the clinical prognosis is not generally considered sufficiently poor to warrant termination and specialist treatment at birth, requiring prenatal planning, and therefore is rarely required. In the autosomal dominant porphyrias, transmission of a severe phenotype is unlikely; indeed, most who inherit the genes for these conditions remain asymptomatic throughout life. However, prenatal diagnosis by DNA analysis of chorionic villous or amniotic cells may be justified in the more severe, and phenotypically predictable, autosomal recessive porphyrias and ‘homozygous’ variants when the family already has a child in whom the pathogenic mutations have been identified. Prenatal diagnosis by molecular analysis has been reported in CEP and HEP.100,101
Methods for mutation detection
Most mutations in the porphyria genes are found in the regions that code for the protein that is the exons or in the areas flanking the exons causing defects in the RNA processing. It is these regions that are targeted for analysis in the first instance. Analysis of the promoter region at the 5′ end of the gene and analysis for the presence of large deletions may also be needed. Other parts of the intronic sequence are not usually analysed.
Identification of a mutation in the proband
The regions of interest are initially amplified from genomic DNA using polymerase chain reaction (PCR) to produce a product that is then used for the investigation. Screening methods may be used to identify parts of the gene that contain variants, for example denaturing high-performance liquid chromatography,102,103 high resolution melting
104
or denaturing gradient gel electrophoresis105,106 before sequencing using dideoxy terminators. As the cost and speed of sequencing improves, screening is becoming less widely used. Direct sequencing of all the regions of interest is now often the preferred option. Because a number of large deletions encompassing one or more exons have been identified in the porphyrias,40–42,107–109 gene dosage analysis by quantitative fluorescent PCR
110
or multiple ligation dependent analysis
40
should always be carried out if a mutation is not detected by sequencing of genomic DNA. Single exon deletions must always be confirmed as variants under primers or probes can mimic these. Confirmation of deletions can be carried out by repeat analysis with different primers or probes or the identification of the breakpoints although the latter may be very laborious. Confirmation of a duplication may be difficult as the size of the duplication may be greater than that easily achieved by long range PCR. This strategy is very similar to that used for a large number of other genes, e.g. BRCA, PMS2, RB1, VHL, ATP7B (
When no mutation can be identified in a patient with biochemically proven porphyria, RNA analysis using a fresh blood sample may be worthwhile. A heterozygous exonic polymorphism will become homozygous in cDNA, or gene dosage 111 analysis will show a 50% reduction in signal, if there is loss of heterogeneity caused by nonsense mediated decay due to a functional intronic mutation.
DNA methods for family screening
When a pathogenic mutation has been identified in a family, analysis of samples from family members is straightforward. The most common method is to sequence the area that contains the mutation although RFLP is used in some centres. 112 Those families with large deletions encompassing primer sites will require dosage analysis or PCR across the breakpoints.
Differentiating a mutation that is causative of porphyria from a harmless sequence variant
As the amount of molecular genetic testing increases more novel sequence variants are identified. Nonsense, frameshift and consensus splice site changes are likely to be pathogenic.
113
However if the pathogenicity of a variant is uncertain guidelines are available from the clinical molecular genetics society (
Annual participation in a specific molecular external quality assessment scheme such as the European Molecular Genetics Quality Network (EMQN: Website;
The European Porphyria Network (EPNET;
Ethical considerations
Consent
The regulations governing genetic testing vary in each country. In September 2011 the Joint Committee on Medical Genetics in the UK published the second edition ‘Consent and confidentiality in genetic practice’. The guidance given in this report was that consent should be obtained prior to a laboratory test with genetic implications being undertaken. It also placed the onus for gaining consent on the clinician requesting the genetic information; so that the laboratory is not required to confirm or document consent.
When a sample is submitted to a laboratory the Joint Committee suggests that the laboratory can assume that consent has been obtained for testing, possible storage and for the use of the sample and the information generated from it to be shared with the members of the donor's family and their health professionals (if appropriate). As a safeguard for the laboratory and to ensure that the patient's wishes are complied with, a customized consent form may be used. One specifically designed for the porphyrias is available on the Cardiff Porphyria Service website
Testing of children
It is recommended that where genetic testing is primarily predictive of an illness or impairment that testing should be delayed until the young person can decide for themselves. However testing may occur when there are specific reasons not to wait until the child is older (Report on the genetic testing of children British Society for Human Genetics 2010). In the acute porphyric disorders it is very rare that acute attacks occur before puberty with only a few documented cases48,114,115 in the world literature. However in order to pre-empt this possibility children in families with acute porphyria, should be offered testing with appropriate consent from parent or guardian. This enables advice on avoidance of precipitating factors to be provided and ensures rapid diagnosis with prompt treatment should an attack occur.
The clinical benefit of genetic diagnosis in EPP is unclear. Presymptomatic diagnosis may be requested in response to parental anxiety. However if a child has not developed symptoms by the age of three years, genetic testing, which is likely to be for carrier status, should be delayed until the child is competent to make an informed decision.
Prenatal or postnatal genetic testing of a sibling in a family with CEP should be carried out so that the child can be protected from light exposure including operating theatre lights during caesarean section and phototherapy for neonatal jaundice.
Conclusions
In summary, the role of genetic testing in the management of patients and their families is not always straight forward and differs among the types of porphyria. In complex cases the advice of a specialist laboratory should be sought.