
Abstract
Objectives:
OTO-201 is a ciprofloxacin otic suspension previously approved by the US Food and Drug Administration to treat children with bilateral otitis media with effusion requiring tympanostomy tube placement. In this phase 3, double-blind, randomized, prospective, sham-controlled, multicenter study, a single dose of OTO-201 was administered to the external auditory canal in subjects with unilateral or bilateral acute otitis externa.
Methods:
Two hundred sixty-two subjects, 3 to 83 years of age, were randomized, and 260 subjects were included in the intent-to-treat analysis population: OTO-201 (0.2 mL, 12 mg, n = 130) or sham (air injection, n = 130). The primary efficacy measure was clinical cure (CC) on day 8, judged by blinded assessor for erythema, edema, otorrhea, and tenderness. Subjects were monitored over 28 days for efficacy and safety.
Results:
OTO-201 demonstrated a significant increase in CC compared with sham at day 8 (69.2% vs 46.1%, P < .001). Higher CC was also noted on day 4 (P = .028), day 15 (P < .001), and day 29 (P < .001). A similar effect was observed in the pathogen-positive population. Single OTO-201 administration in the office setting was well tolerated by subjects.
Conclusions:
In this study in subjects with acute otitis externa, a single administration of 12 mg OTO-201 to the external auditory canal demonstrated a significantly higher proportion of subjects with CC and bacterial eradication compared with sham starting on day 4 and on all other observation days through day 29, with no safety or tolerability concerns identified. OTO-201 is the first agent in a randomized phase 3 study to demonstrate the efficacy and safety of a single-dose, health care professional–administered topical antibiotic for the treatment of acute otitis externa.
Keywords
Introduction
Acute otitis externa (AOE), also known as “swimmer’s ear,” is a common condition in pediatric and adult populations characterized by diffuse inflammation of the external auditory canal (EAC). On the basis of 2007 data from the Centers for Disease Control and Prevention, approximately 1 in 123 persons in the United States visited ambulatory care centers and emergency departments because of AOE, equating to 2.4 million patients. Incidence of AOE is shown to peak during the summer months, regionally in the South, and in children between 5 and 14 years of age. 1 Per the clinical practice guidelines published by the American Academy of Otolaryngology–Head and Neck Surgery Foundation (AAO-HNSF), diagnosis of AOE requires a rapid onset (generally within 48 hours) in the past 3 weeks of signs and symptoms of EAC inflammation.2,3 Symptoms include otalgia, itching, and aural fullness. Signs include tenderness of the tragus and pinna, diffuse ear canal edema, conductive hearing loss, erythema, and otorrhea. Overall, 90% of AOE cases are unilateral; in North America nearly all of the time (98%), AOE is caused by bacterial infection. 3 Pseudomonas aeruginosa and Staphylococcus aureus are the most common pathogens responsible for AOE, with Staphylococcus species being less prevalent in children than in adults. 4
Major components of managing AOE include cleaning the EAC, addressing pain control, and treating the infection with a topical antibiotic. Removal of cerumen, desquamated skin, and purulent discharge from the EAC improves healing and enhances the penetration of topical therapies to the site of infection. Current standard of care, consistent with AAO-HNSF guidelines, recommends the use of topical antibiotics, as opposed to systemic antibiotics, for the treatment of diffuse, uncomplicated AOE. 3 A variety of topical preparations are approved by the US Food and Drug Administration (FDA) for treating AOE. These agents incorporate, either as monotherapy or combination therapy, an antibiotic (such as aminoglycoside, polymyxin B, quinolones, or combination agents), a corticosteroid, or a low-pH antiseptic. Per the guidelines, no meaningful differences in clinical outcomes have been noted on the basis of class of drug, use of a quinolone versus nonquinolone preparation, or for monotherapy versus combination drugs with or without a steroid. 3
A limitation associated with all currently FDA-approved topical agents for treating AOE is the requirement of multiple applications (up to 4 times/day) over multiple days (usually over 7 days) by the patient or caregiver.5-7 The practice guideline recognizes multiple challenges associated with the administration of topical drug delivery to an infected external ear canal, citing poor patient adherence to therapy, difficulties with application (ie, “missing” the ear canal), presence of debris in the canal, and edema that leads to closing of the EAC. 3 Furthermore, the guidelines state that difficulties with self-administration of topical agents arise because it must be done by “feel.” Indeed, data from a blinded study in 39 adult patients with AOE prescribed otic drops showed that only 40% of patients who self-medicated did so appropriately during the first 3 days, with a tendency often to undermedicate. 8
OTO-201 was designed to be a single-dose, health care professional (HCP)–administered formulation of ciprofloxacin suspended in a thermosensitive and thermoreversible poloxamer (P407). 9 When OTO-201 is exposed to the temperature within the ear, it quickly transitions from a liquid to a gel. P407 serves as a novel drug “carrier,” allowing suspended microparticles of ciprofloxacin to be deposited and solubilized over time. OTO-201 was previously approved by the FDA to treat children with bilateral otitis media with effusion requiring tympanostomy tube placement. 9 A prior phase 2 clinical study in AOE demonstrated the feasibility of delivering 0.2 mL of OTO-201 in children and adult subjects, with >90% of HCPs reporting that it was “very easy” to administer the treatment in an outpatient, ambulatory care setting when using readily available catheters. 10 In this phase 3 clinical study we evaluated the efficacy, safety, and tolerability of a single administration of OTO-201 to the EAC in subjects presenting with AOE.
Methods
Study Design
This was a double-blind, randomized, prospective, sham-controlled, multicenter phase 3 clinical study of OTO-201 (6% ciprofloxacin otic suspension; OTIPRIO; Otonomy, Inc., San Diego, California, USA) administered as a single dose in the outpatient, office-based setting to children and adults with AOE. Institutional review board (IRB; Schulman Associates IRB [IRB00000971], Cincinnati, Ohio, USA; AltaMed Health Services Corporation [FWA00002187]; Emory IRB [IRB00000569]; Vanderbilt University IRB [IRB00000475]; Medical University of South Carolina IRB [IRB00000027]; and Saint Louis University IRB [FWA00005304]) approval and written informed consent were obtained prior to study-related procedure initiation. The study was conducted in accordance with the regulations and guidelines of the FDA, the Declaration of Helsinki, and the International Conference on Harmonization Good Clinical Practice guidelines.
Key inclusion criteria required that subjects be male or female, be 6 months of age or older, have a clinical diagnosis of unilateral or bilateral AOE, and have a combined numeric severity score of ≥4 in at least 1 affected ear at the screening visit for tenderness, erythema, and edema (ie, each measure was scored 0 = none [complete absence of any signs or symptoms], 1 = mild [slight], 2 = moderate [definitely present], or 3 = severe [marked, intense]). In subjects with bilateral AOE, only 1 ear must have met this criterion, and both ears were assessed, cultured, and treated with OTO-201 or sham. Key exclusion criteria were the following: history of allergy to ciprofloxacin, known tympanic membrane perforation in either ear, severe otitis externa (OE) that either included auricular cellulitis or chondritis or prevented administration of OTO-201, chronic OE, defined as either having 1 or more previous episodes of OE within the past 3 months or more than 3 previous episodes of OE within the past year, eczematoid OE, or fungal OE. Subjects with baseline cultures growing group A Streptococcus were excluded from efficacy analysis and treated with standard of care, including systemic therapy as needed. A complete list of the inclusion and exclusion criteria is provided in the Supplemental Section.
Randomization and Study Intervention
OTO-201 (12 mg delivered in 0.2 mL) was evaluated relative to sham (air injection) using 1:1 randomization. A permuted block randomization algorithm schedule was used, and the randomization process was deployed using a Web-based interactive response system. Eligible subjects were randomized to treatment assignment on day 1 to OTO-201 or sham. Administration of OTO-201 or sham was conducted by an unblinded investigator. Treatment syringes were prepared by an unblinded qualified medical professional or the unblinded investigator. Each syringe was prepared according to detailed instructions in a manner that prevented the visualization of syringe contents by all other study staff members through the use of a syringe overlabel. Any interaction with subjects with regard to the collection, review, or discussion of study assessments was done by the study coordinator, blinded investigator, or someone other than the qualified medical professional who prepared the syringe and the unblinded investigator who administered OTO-201 or sham. HCPs who administered study drug were not blinded, because of the visual difference during administration, and were instructed not to discuss any potential visual differences observed in study material with subjects or study staff members. Prior to administration, the ear canal was to be cleaned of all fluid and debris (ie, dry mop or suctioning). OTO-201 was administered via blunt catheter slowly into the EAC (injected into the canal with the syringe tip advanced medially approximately 5 mm beyond the cartilaginous junction), and the catheter was slowly withdrawn while injecting. During the study, antibiotics other than OTO-201 were discouraged; however, if deemed necessary for the welfare of the subject, topical antibiotics (eg, lotions, creams, ointments) could have been administered. Concomitant use of otic antibiotics and/or otic drops of any kind was prohibited.
Study Outcomes
Otoscopic examinations were used to assess the signs (edema, erythema) and symptoms (tenderness) of OE by a blinded investigator in both ears at every study visit using the scoring scale used during study entry: 0 = none (complete absence of any signs or symptoms), 1 = mild (slight), 2 = moderate (definitely present), 3 = severe (marked, intense). In addition, the presence and characterization of otorrhea were documented for all study visits. Otorrhea was defined as trace (tympanic membrane is moist in appearance, with no fluid and/or no accumulation of otorrhea present in the EAC), moderate (accumulation of otorrhea that has not obscured the visualization of the EAC), or copious (accumulation of otorrhea that has obscured the visualization of the EAC).
A swab of the EAC from each affected ear was taken for microbiologic culture and susceptibility prior to study drug or sham administration (day 1) and at subsequent visits if edema, erythema, tenderness, or otorrhea was observed by the blinded assessor. If culture positive, microorganisms were identified by using a Bruker (Billerica, Massachusetts, USA) matrix-assisted laser desorption ionization time-of-flight Biotyper (only on isolates identified as S aureus, Moraxella catarrhalis, Streptococcus pneumoniae, Haemophilus influenzae, P aeruginosa, or Pseudomonas otitidis) performed at a central laboratory.
Subjects or their caregivers reported assessment of otalgia in the affected ear in a daily diary using the Wong-Baker FACES Pain Rating Scale ranging from 0 to 10 (0 = no hurt, 10 = hurts worst) on days 1 through 15. For subjects whose ear pain ended during days 1 through 15, the date when the ear pain ended was recorded. The daily diary was completed only for subjects mature enough to provide appropriate responses to their levels of otalgia, typically 3 years or older. Any subject who did not show improvement by the day 8 follow-up visit or whose daily diary indicated no improvement in otalgia was provided the standard of care for AOE at the discretion of the investigator. Safety assessments included adverse events (AEs), vital sign measurements, otoscopic examinations, and concomitant medications.
The primary endpoint was the proportion of subjects with clinical cure (CC) on day 8. CC was defined as complete resolution of signs and symptoms (ie, tenderness, erythema, otorrhea, and edema, as determined by the blinded investigator) with no concomitant antibiotics (systemic or topical given in the study ear) given for any reason at or prior to the study visit with complete resolution of signs and symptoms. Complete resolution of signs and symptoms was achieved when scores for tenderness, erythema, otorrhea, and edema were equal to zero at the assessment day. Other efficacy endpoints evaluated were CC on days 4, 15, and 29 as determined by blinded investigator; microbiologic eradication on days 4, 8, 15, and 29; the proportion of subjects with cessation of otorrhea; and time to cessation of otalgia.
Statistical Analysis
The intent-to-treat (ITT) analysis set included all subjects who were randomized and did not have cultures positive for group A Streptococcus cultured on day 1. The microbiologic ITT (mic-ITT; ie, the pathogen-positive population) included all ITT subjects with positive baseline cultures for either P aeruginosa or S aureus. The primary efficacy endpoint was compared between the treatment groups at a .05 level of significance using the ITT analysis set. The proportion and corresponding Clopper-Pearson 95% confidence intervals for subjects demonstrating CC were presented by treatment group and study visit (through days 4, 8, 15, and 29). Differences in proportions and corresponding exact 95% confidence intervals were also presented. Treatment comparisons with regard to proportions were performed using the Fisher exact test. Once the primary objective was met in the ITT analysis set, the secondary endpoints were compared between the treatment groups in the ITT analysis set. To control the family-wise type I error for the evaluation of the secondary endpoints, a gatekeeping strategy (sequential closed testing procedure) was implemented. The following sequence specified the steps: CC day 15 using ITT analysis set, CC day 8 using mic-ITT analysis set, CC day 15 using mic-ITT analysis set, CC day 4 using mic-ITT analysis set, CC day 4 using ITT analysis set, and time to cessation of otalgia using ITT analysis set.
Results
Subjects and Baseline Characteristics
Two hundred sixty-two subjects were randomized between June and November 2016 at 37 clinical sites in the United States (Figure 1). Two randomized subjects (both in the sham group) tested positive for group A Streptococcus and completed the study. Per the protocol, both subjects were excluded from the efficacy analysis but included in the safety population (Figure 1). Clinical sites included both ear, nose, and throat (ENT) providers (16 sites) and non-ENT providers (21 sites, including pediatrician offices, primary care physician offices, urgent care centers, and outpatient centers). Table 1 summarizes subjects’ baseline demographics and disease characteristics. Otorrhea was present at baseline in the study ear of 59.2% of subjects in the ITT population. The predominant type of otorrhea in the study ear at baseline in those subjects who presented with otorrhea was purulent (37.3% of subjects). The mean combined signs and symptoms score of study ear was similar between the treatment groups (sham, 6.2; OTO-201, 6.3; Table 1). Rates of edema, erythema, and tenderness in the study ear at baseline were 98.5%, 97.7%, and 99.2%, respectively. Baseline scores were similar for subjects at ENT sites (mean score, 6.3; n = 81) and non-ENT sites (mean score, 6.2; n = 179). A total of 109 subjects (41.9% of the ITT population) had positive cultures in the study ear at baseline; the most common organisms cultured from the study ear were P aeruginosa (29.6% of subjects) and S aureus (16.9% of subjects).
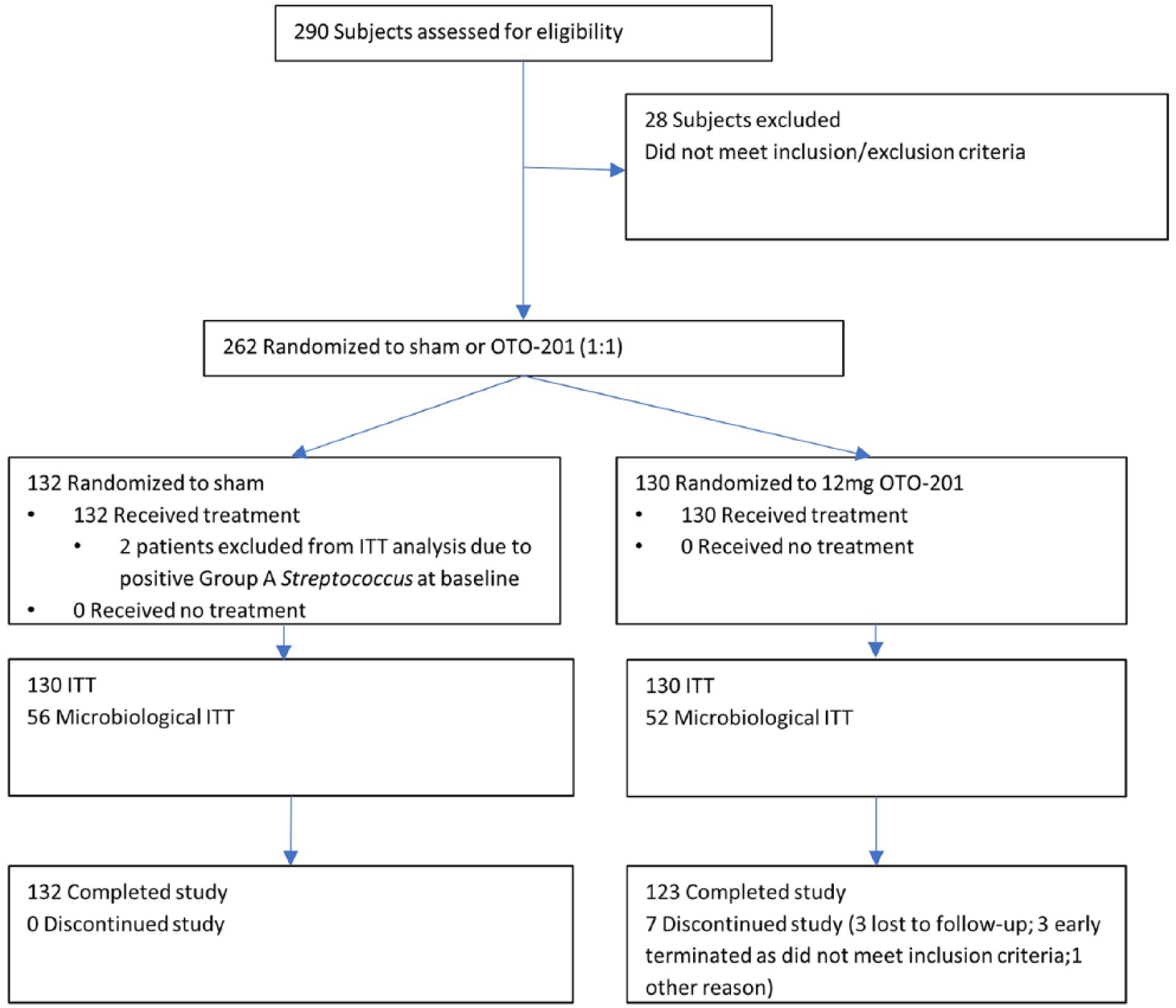
Consolidated Standards of Reporting Trials diagram indicating flow of study subjects.
Baseline Demographics and Disease Characteristics (Intent-to-Treat Population). a
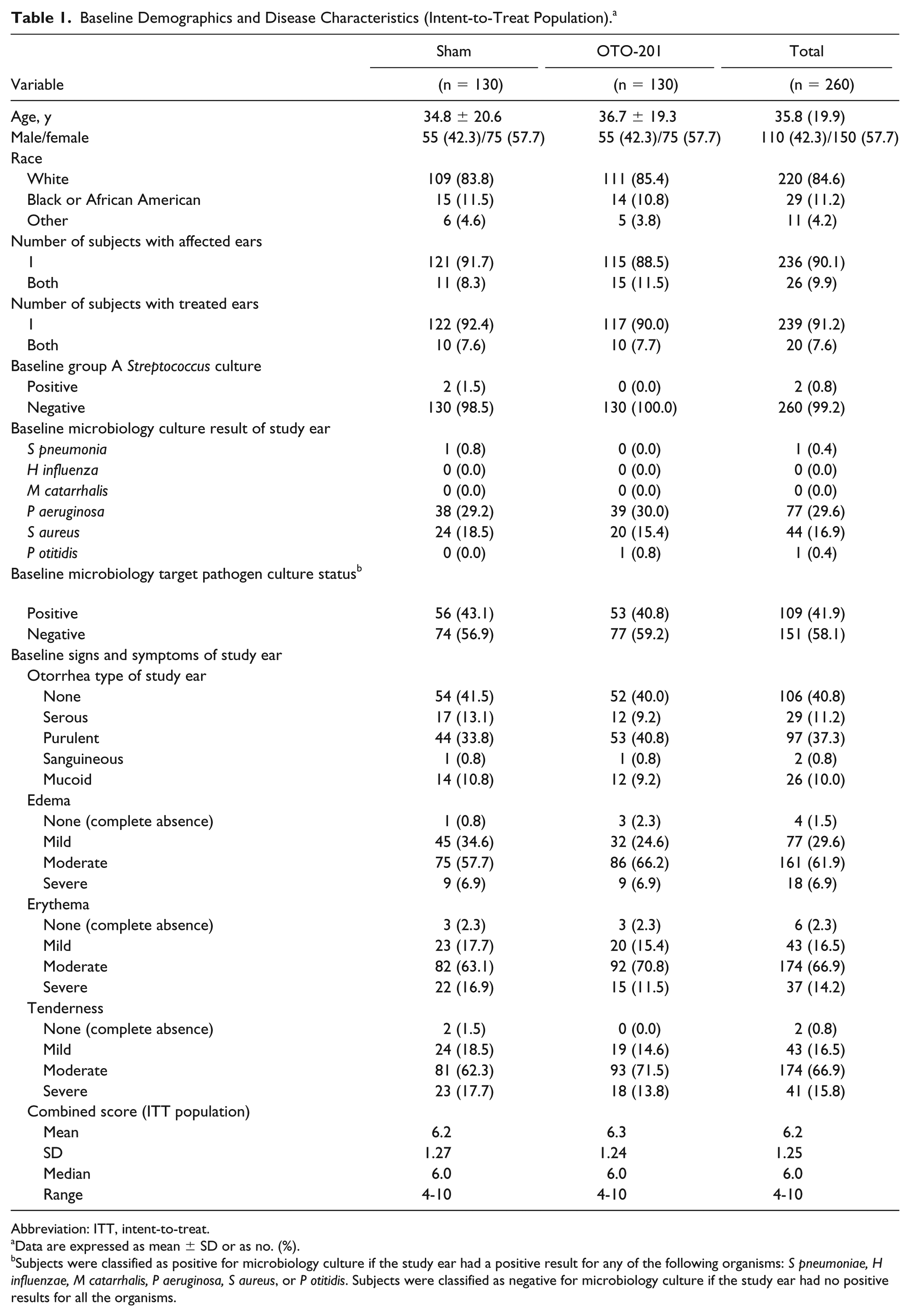
Abbreviation: ITT, intent-to-treat.
Data are expressed as mean ± SD or as no. (%).
Subjects were classified as positive for microbiology culture if the study ear had a positive result for any of the following organisms: S pneumoniae, H influenzae, M catarrhalis, P aeruginosa, S aureus, or P otitidis. Subjects were classified as negative for microbiology culture if the study ear had no positive results for all the organisms.
Efficacy
The primary endpoint (proportion of subjects with CC on day 8) demonstrated a significant increase in the OTO-201 group compared with sham (90 of 130 [69.2%] vs 60 of 130 [46.1%]; difference in proportion, 23.1%; 95% confidence interval, 10.7%-34.6%; P < .001) (Figure 2). For the secondary endpoints, statistical significance was achieved for CC through day 15 using the ITT analysis set (difference in proportion, 23.8%; P < .001), CC on day 8 using the mic-ITT analysis set (difference in proportion, 25.7%; P = .012) (Figure 3), and CC on day 15 using the mic-ITT analysis set (difference in proportion, 35.2%; P < .001). Statistical significance was not achieved for CC on day 4 using the mic-ITT analysis set (difference in proportion, 9.5%; P = .291). CC on day 4 using the ITT analysis set (difference in proportion, 13.8%; P = .028) followed CC on day 4 using the mic-ITT in the gatekeeping sequential testing procedure criteria, and therefore the P value should be considered nominal. For the primary endpoint (CC on day 8), OTO-201 results were similar between ENT (66% CC) and non-ENT sites (71% CC). During the study, the number of OTO-201 subjects who received antibiotic standard of care through day 8 was 12 (9%), and the number of sham subjects was 42 (32%).
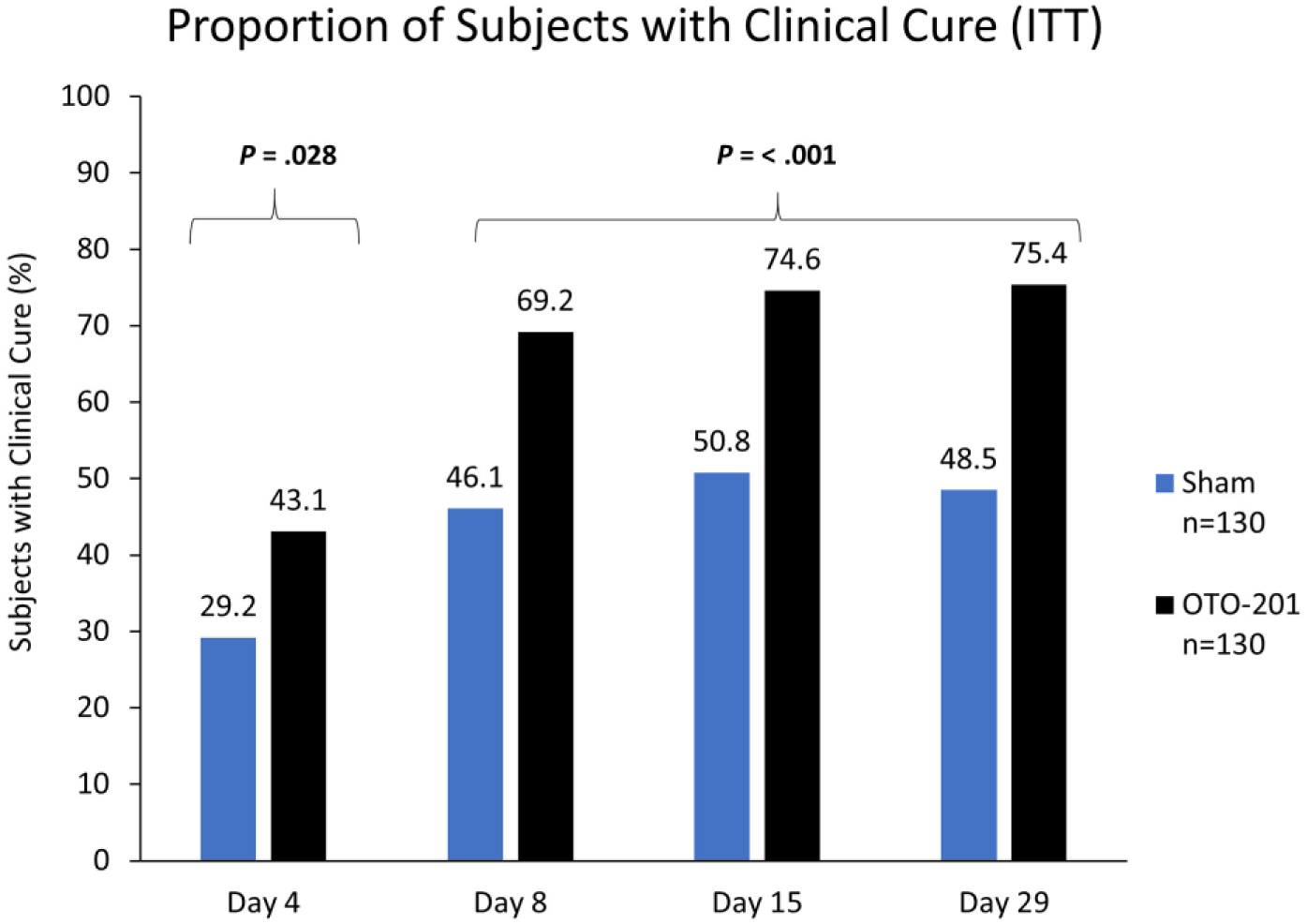
Clinical cure rates on days 4 to 29.
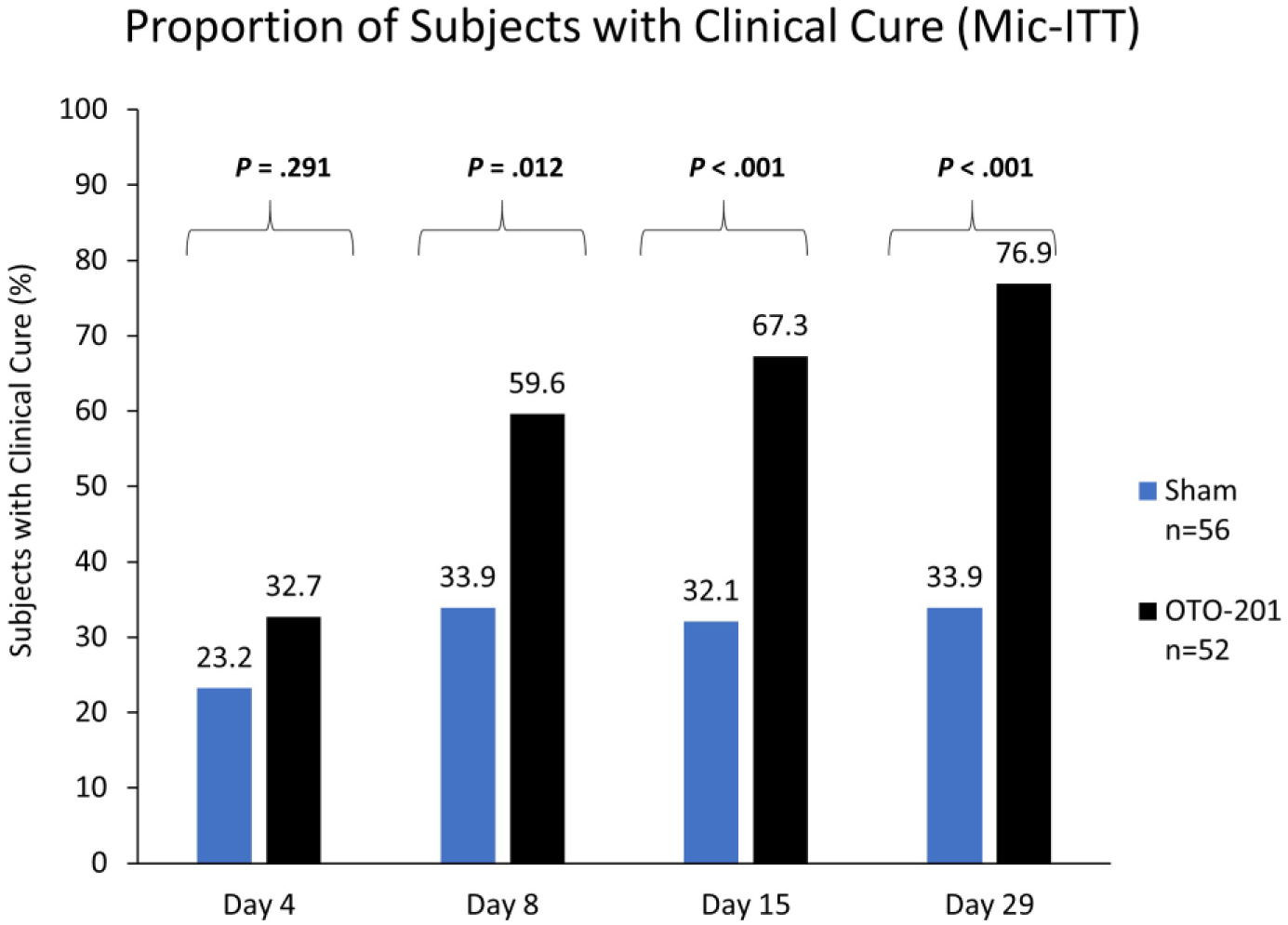
Clinical cure rates in the microbiologic culture–positive group on days 4 to 29.
For the time through day 4, the proportion of subjects with cessation of otorrhea was greater in the OTO-201 group than the sham group (58.6% vs 42.1%; Table 2). The proportion of subjects with cessation of otorrhea was greater in the OTO-201 group than the sham group on days 8, 15, and 29 (differences in proportion, 25.0% through day 8, 29.3% on day 15, and 37.8% on day 29). Median time to cessation of otalgia through day 15 was 8.0 and 11.0 days for the OTO-201 group and sham group, respectively (P = .008).
Proportion of Subjects With Cessation of Otorrhea (Intent-to-Treat Population). a
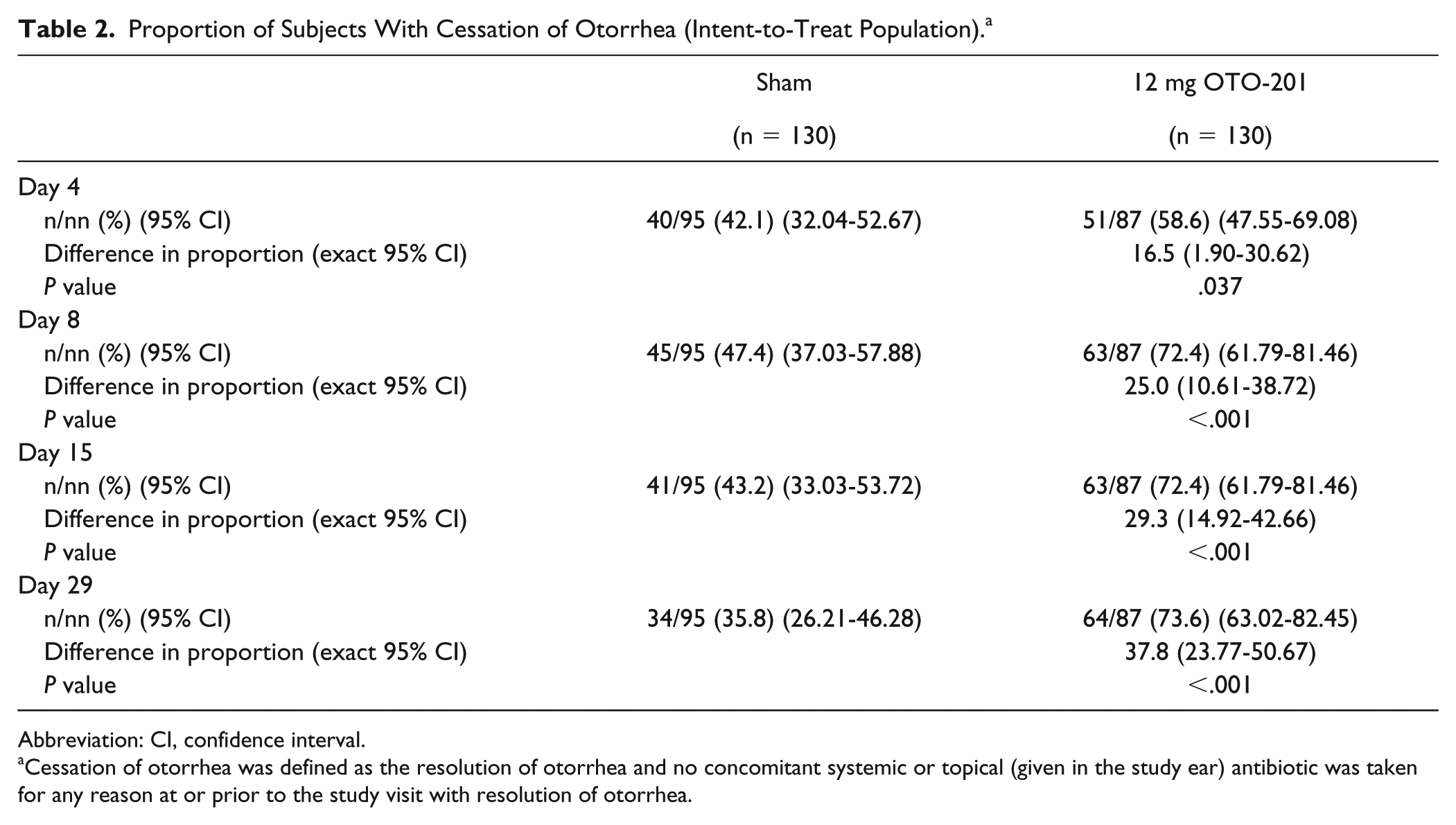
Abbreviation: CI, confidence interval.
Cessation of otorrhea was defined as the resolution of otorrhea and no concomitant systemic or topical (given in the study ear) antibiotic was taken for any reason at or prior to the study visit with resolution of otorrhea.
The proportion of subjects with microbiologic eradication on day 8 was greater in the OTO-201 group than in the sham group (76.9% vs 37.5%; difference in proportion, 39.4%; 95% confidence interval, 21.3%-55.9%; P < .001) (Table 3).
Microbiologic Eradication Results by Culture at Baseline and Following Treatment (Microbiologic Intent-to-Treat Population) a

Abbreviation: CI, confidence interval.
The microbiologic intent-to-treat set included all intent-to-treat subjects with positive baseline cultures for either P aeruginosa or S aureus.
Safety
The safety analysis population comprised all subjects who received study drug (n = 259). A total of 54 subjects (20.8%) reported 82 treatment-emergent AEs during the study (Table 4). The proportion of subjects reporting treatment-emergent AEs was similar in the OTO-201 group and the sham group (18.9% vs 22.7%, respectively). No deaths, serious AEs, or treatment-emergent AEs leading to subject discontinuation were reported. Treatment-emergent AEs reported in 2 or more patients in the OTO-201 group and greater than the sham group were headache (2.4% and 0.8%), ear pruritus (2.4% and 1.5%), ear pain (2.4% and 2.3%), ear discomfort (1.6% and 0%), nasal congestion (1.6% and 0%), and otitis media (1.6% and 0.8%). Otoscopic examination observations (cerumen, middle ear, and tympanic membrane) were similar between the OTO-201 and sham groups throughout the study.
Most Frequently Reported TEAEs (≥2% in Any Treatment Group) (Safety Population). a
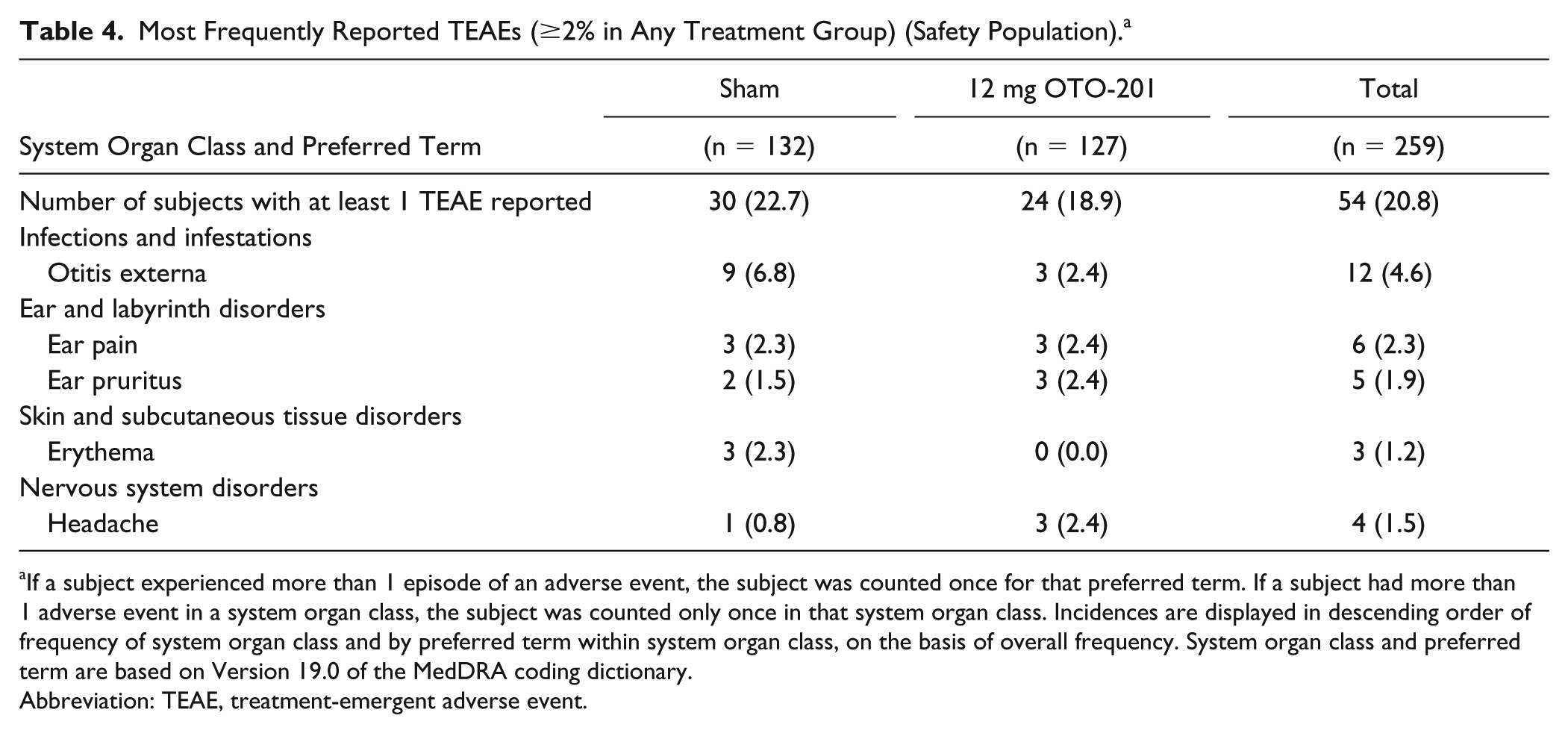
If a subject experienced more than 1 episode of an adverse event, the subject was counted once for that preferred term. If a subject had more than 1 adverse event in a system organ class, the subject was counted only once in that system organ class. Incidences are displayed in descending order of frequency of system organ class and by preferred term within system organ class, on the basis of overall frequency. System organ class and preferred term are based on Version 19.0 of the MedDRA coding dictionary.
Abbreviation: TEAE, treatment-emergent adverse event.
Discussion
AOE is diagnosed clinically on the basis of signs and symptoms of EAC inflammation. In the present study, more than 97% of subjects presented at baseline with the classic signs and symptoms of edema, erythema, and tenderness. Given the bacterial origin for AOE and high local antibiotic concentrations achieved with topical antibiotic therapies, the AAO-HNSF practice guidelines for AOE recommend the use of topical antibiotic therapy while strongly recommending against prescribing systemic antibiotics as initial therapy for diffuse, uncomplicated AOE. This phase 3 study evaluated the efficacy of a single topical administration of OTO-201 for the treatment of AOE. The proportion of subjects meeting the primary endpoint (proportion of subjects achieving CC on day 8) was greater and statistically significant in the OTO-201 group compared with the sham group (69% vs 46%, a 50% relative increase). Using a statistical gatekeeping strategy to control family-wise type I error, statistical significance was achieved for multiple secondary endpoints including CC on day 15 using the ITT analysis set and CC on day 8 and 15 using the Mic-ITT analysis set. All other statistical comparisons were considered exploratory but consistently showed treatment group differences favoring OTO-201 over sham. Cessation of otorrhea showed treatment group differences in favor of OTO-201 over sham for all visits. OTO-201 treatment also resulted in greater microbiologic eradication than sham (Mic-ITT population) through days 4, 8, 15, and 29 and a shorter median time to cessation of otalgia through day 15.
These findings demonstrate that a single administration of 0.2 mL (12 mg) OTO-201 provided greater resolution of the signs and symptoms of AOE compared with sham control. The CC rate with a single dose in this trial on day 8 (69.2%) is consistent with the reported rates of 65% to 80% seen by day 10 with existing antibiotic topical therapies given over multiple days. 11 Although a weakness of this trial is the lack of an active antibiotic comparator as a third arm, such a trial would be problematic, because subject and investigator blinding cannot be maintained and treatment bias is likely. Moreover, topical treatments containing ciprofloxacin are already known to be effective in the treatment of AOE (while conclusive data from robust clinical trials are still needed to determine whether the addition of steroids to topical quinolone antibiotics provides earlier relief of AOE symptoms).12,13 In addition, this study excluded subjects with atypical, severe, and chronic OE, and therefore the safety and efficacy of OTO-201 are unknown in treating severe and chronic OE. Similar to other topical therapies for AOE, OTO-201 should not be administered alone without systemic antibiotics for subjects who present with severe AOE.
The results seen with OTO-201 are notable because they are the first to demonstrate CC and bacterial eradication in AOE resulting from a single-dose topical ciprofloxacin suspension administered by HCPs in an office setting. OTO-201 ensures compliance by eliminating the need for the subject to self-administer the antibiotic after the visit, numerous times a day over 7 days, as currently required with existing FDA-approved topical antibiotic therapies. Data from England et al 8 showed poor patient compliance when self-administering multidose antibiotic ear drops, with a tendency often to undermedicate and only 43% of patients appropriately self-medicating topical therapies over 7 days. Current AAO-HNSF practice guidelines for AOE further validate that treatments that involve less frequent dosing schedules and a shorter duration lead to higher patient treatment acceptability. 3 In the study, both physicians and other HCPs (registered nurses, nurse practitioners, licensed vocational nurses, physician assistants, and audiologists) administered OTO-201. The HCPs were able to administer the full 12-mg (2-mL) dose of OTO-201 with either suction needles or Angiocath catheters, which are commonly found in the outpatient setting.
Future researchers may want to explore through registries the impact OTO-201 may have on increasing the percentage of patients with AOE who are prescribed topical antibiotic preparations (as opposed to systemic antibiotics), as this is now part of the Centers for Medicare and Medicaid Services–mandated Physician Quality Reporting System quality measure for AOE. Additionally, given the number of treatment options available for patients with AOE, a greater understanding of priorities that ultimately affect patient and physician treatment preferences would be valuable.
Conclusions
This double-blind, randomized, prospective, controlled, multicenter study phase 3 clinical study demonstrated that a single administration of 12 mg OTO-201 resulted in a significantly higher proportion of subjects with CC and bacterial eradication compared with sham in children and adults with AOE at all prespecified time points. OTO-201 treatment was well tolerated by patients. Administration of OTO-201 eliminated concerns with patient compliance and was accomplished by ENTs and non-ENT HCPs in the primary care outpatient setting. Findings from this trial support the use of OTO-201 as a single-dose treatment option administered by HCPs for patients with AOE.
Supplemental Material
Suppl_OTO-201_P3_AOE_AORL – Supplemental material for OTO-201 for the Treatment of Acute Otitis Externa: Results from a Phase 3 Randomized Clinical Study
Supplemental material, Suppl_OTO-201_P3_AOE_AORL for OTO-201 for the Treatment of Acute Otitis Externa: Results from a Phase 3 Randomized Clinical Study by John Ansley, Eric A. Mair, Hamid Namini, Chung H. Lu and Carl LeBel in Annals of Otology, Rhinology & Laryngology
Footnotes
Acknowledgements
We thank the subjects for their participation in this study, the clinical study site staff members, and the Rho members of the study team. We express our sincere gratitude to the following principal investigators who participated in the study: Aftab Ahmad, MD, Research Trials Worldwide, Humble, Texas; Rajasekaran Annamalai, MD, East Montgomery County Clinic, Splendora, Texas; John Ansley, MD, Carolina Ear, Nose and Throat Clinic, Orangeburg, South Carolina; Eddie Armas, MD, Well Pharma Medical Research Corporation, South Miami, Florida; Michael Armstrong, MD, Richmond Ear, Nose & Throat, Richmond, Virginia; Paul Beckett, MD, Elite Clinical Trials, Blackfoot, Idaho; Matthew Brown, MD, Iowa Head & Neck PC, Des Moines, Iowa; Tami Bruce, MD, Central Arizona Medical Associates PC/Radiant Research, Mesa, Arizona; James Cain, MD, Family Medicine Clinic, Lampasas, Texas; Alejandro Cordero, MD, Miami Dade Medical Research Institute LLC, Miami, Florida; Hector Cordero, MD, Pharma Research International, Naples, Florida; Steven Davis, MD, Breathe Clear Institute, Torrance, California; Patrick Dennis, MD, DelRicht Research, New Orleans, Louisiana; Franklin Douglas, MD, Conroe and Spring, Texas; Ann Edmunds, MD, Omaha Ear, Nose & Throat, Omaha, Nebraska; Dale Robert Ehmer, MD, Ear, Nose & Throat Associates of Texas, McKinney, Texas; Robert Florea, MD, Buckeye Health & Research, Columbus, Ohio; Augusto Focil, MD, Diverse Research Solutions, Oxnard, California; Darin Gregory, MD, Pioneer Clinical Research LLC, Bellevue, Nebraska; Jonathan Hubbard, MD, Chrysalis Clinical Research, St. George, Utah; Matthew Hummel, MD, Fountain Hills Family Practice PC/Radient Research, Fountain Hills, Arizona; Janice Johnston, MD, Arrowhead Health Centers, Glendale, Arizona; Najmuddin Karimjee, MD, Tidwell Medical Center, Houston, Texas; Brian Keith MacGillivray, MD, DCT-Stone Oak LLC/Discovery Clinical Trials, San Antonio, Texas; Audrey Lee Jones, MD, DCT-McAllen Primary Care Research LLC/Discovery Clinical Trials, McAllen, Texas; Eric A. Mair, MD, Charlotte Eye, Ear, Nose, and Throat Associates, Charlotte, North Carolina; John McClay, MD, Frisco ENT for Children, Frisco, Texas; Lorely Mendez, MD, San Marcus Research Clinic, Miami, Florida; Misal Khan, MD, Panama City, Florida; Jonathan Moss, MD, Charlotte Eye, Ear, Nose & Throat Associates, Matthews, North Carolina; Robert Scott, MD, White Oak Family Physicians PA/Asheboro Research Associates, Asheboro, North Carolina; Anthony Mikulec, MD, St. Louis University, St. Louis, Missouri; John Howard Taylor, MD, Biosolutions Clinical Research Center, La Mesa, California; Cynthia Wellman, MD, Ear, Nose & Throat Associates, Fort Wayne, Indiana; Jeffrey Rosenbloom, MD, Alamo ENT, San Antonio, Texas; and Donald Welsh, MD, Advanced ENT & Allergy, Louisville, Kentucky.
Authors’ Note
This research was the subject of an oral presentation by John Ansley, MD, at the annual meeting of the American Academy of Otolaryngology–Head and Neck Surgery, September 11, 2017, Chicago, Illinois.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Ansley’s institution received clinical study funding from Otonomy, Inc. Dr. Mair’s institution received clinical study funding from Otonomy, Inc. Dr. Namini is a former employee and shareholder of Otonomy, Inc. Dr. Lu is a former employee and shareholder of Otonomy, Inc. Dr. LeBel is a former employee and shareholder of Otonomy, Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by Otonomy, Inc.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.