
Abstract
The mesentery is a common site of metastasis from gastrointestinal, pancreatic, and biliary cancers. Primary mesenteric cancers are rare and usually mesenchymal and benign. Mesenteric leiomyosarcoma is a rare, malignant smooth muscle sarcoma with an incidence of 1:350000. It usually arises from the vasculature of the mesentery. The ileum of the small bowel is the most common site of origin. Due to its low incidence, preoperative diagnosis is difficult. This is a report of a 71-year-old woman who presented with several months of a lower abdominal mass and recent onset of associated abdominal discomfort. An earlier colonoscopy 8 months previously was unremarkable. A recent abdominal and pelvic computed tomography scan revealed a necrotic mass in the central mesentery. She underwent surgical resection of the mass to include the overlying segment of the small intestine and had an uneventful convalescence. Mesenteric leiomyosarcoma is a rare tumor that is diagnosed based on histological examination with immunohistochemistry. As a result, there is minimal information on its clinical presentation, pathology, and treatment.
Leiomyosarcomas (LMSs) are soft tissue sarcomas that present with an aggressive clinical course. A 71-year-old well-appearing woman reported that she developed a palpable, enlarging lower abdominal mass over a period of several months. A week before presentation, she additionally experienced gradual periumbilical tenderness, dull pain, bloating, fullness, dyspepsia, and a loss of urinary control. No weight gain or weight loss. A colonoscopy performed 8 months prior was unremarkable. A computed tomography (CT) scan of the abdomen and pelvis revealed a large lobulated necrotic mass within the central mesentery, just below the level of the umbilicus measuring 8.2 × 5.5 × 7.4 cm (Figure 1). The peripheral aspect of the mass demonstrated heterogeneous enhancement and displaced adjacent bowel loops. There was no evidence of adenopathy or distant metastasis.
The patient underwent a midline laparotomy. There was no evidence of peritoneal metastatic disease. A large mobile mid-small bowel mesenteric mass with associated loops of the small intestine was resected. The postoperative course was uneventful. The tumor specimen consisted of an 85.0 cm in length segment of tortuous small bowel that was partially adherent to a 9.0 × 7.0 cm mesenteric mass. The mass was grossly intact with patchy necrosis and areas of hemorrhage. The mass extended to within 0.5 cm of the closest mesenteric margin and grossly involved the wall of the small bowel. The tumor narrowed the small intestine, but no gross extension into the lumen was noted. There was involvement of the serosal surface, with marked roughening and adhesions noted on the overlying surface and mesentery. The small bowel margin, however, was negative, with folded pink-tan, unremarkable mucosa free of lesions.
The mass was classified as a grade 3 LMS, as defined by marked cellularity, anaplasia, and a mitotic index >10/10 high-power fields (HPFs). Immunohistochemistry demonstrated positivity for desmin, but no staining for the markers associated with a gastrointestinal stromal tumor (GIST) such as CD34, CD117, and delay of germination 1 (DOG1). The tumor was also negative for S-100 and displayed a high mitotic activity of 25/10 HPFs. The patient did not receive any postoperative chemotherapy or radiation.
Many smooth muscle tumors previously reported as LMS before the introduction of GIST were later reclassified as GIST. Both tumors are similar microscopically and grossly. 1 Most smooth muscle tumors of the gastrointestinal tract are GISTs, which are defined by immunohistochemical (IHC) positive staining for KIT, CD34, CD117, DOG1, and sometimes by molecular evaluation of activating mutations in the KIT or Platelet-derived growth factor receptor alpha (PDGFRA) genes. LMS can be differentiated from GIST through positive IHC staining for desmin, smooth muscle actin, vimentin, and h-caldesmon, and negativity for GIST markers. Histologically, LMS is characterized by intersecting, sharply marginated fascicles of spindle cells with elongated, hyperchromatic nuclei and abundant eosinophilic cytoplasm. Varying degrees of pleomorphism can occur. 1 LMS can be differentiated from leiomyoma by the presence of alternating areas of necrosis and hemorrhage.
Effective treatment strategies for intestinal LMS are unavailable due to its rarity. Surgical resection with a wide margin of normal surrounding tissue is the mainstay of therapy. LMS responds poorly to chemoradiation therapy, but new therapies are currently being investigated. 2
The late presentation of LMS and its aggressive mitotic activity leads to a high malignant potential and a poor prognosis. Currently, diagnosis is confirmed through pathology and immunohistochemical staining. There is an overall 5-year survival rate of 20-30%.
3
Mortality is typically due to distant metastasis, most commonly to the liver. Better prognoses will be attainable in the future as we learn more about this rare tumor Figure 1. Longitudinal computed tomography image of a leiomyosarcoma that is adherent to the small intestine without invading it.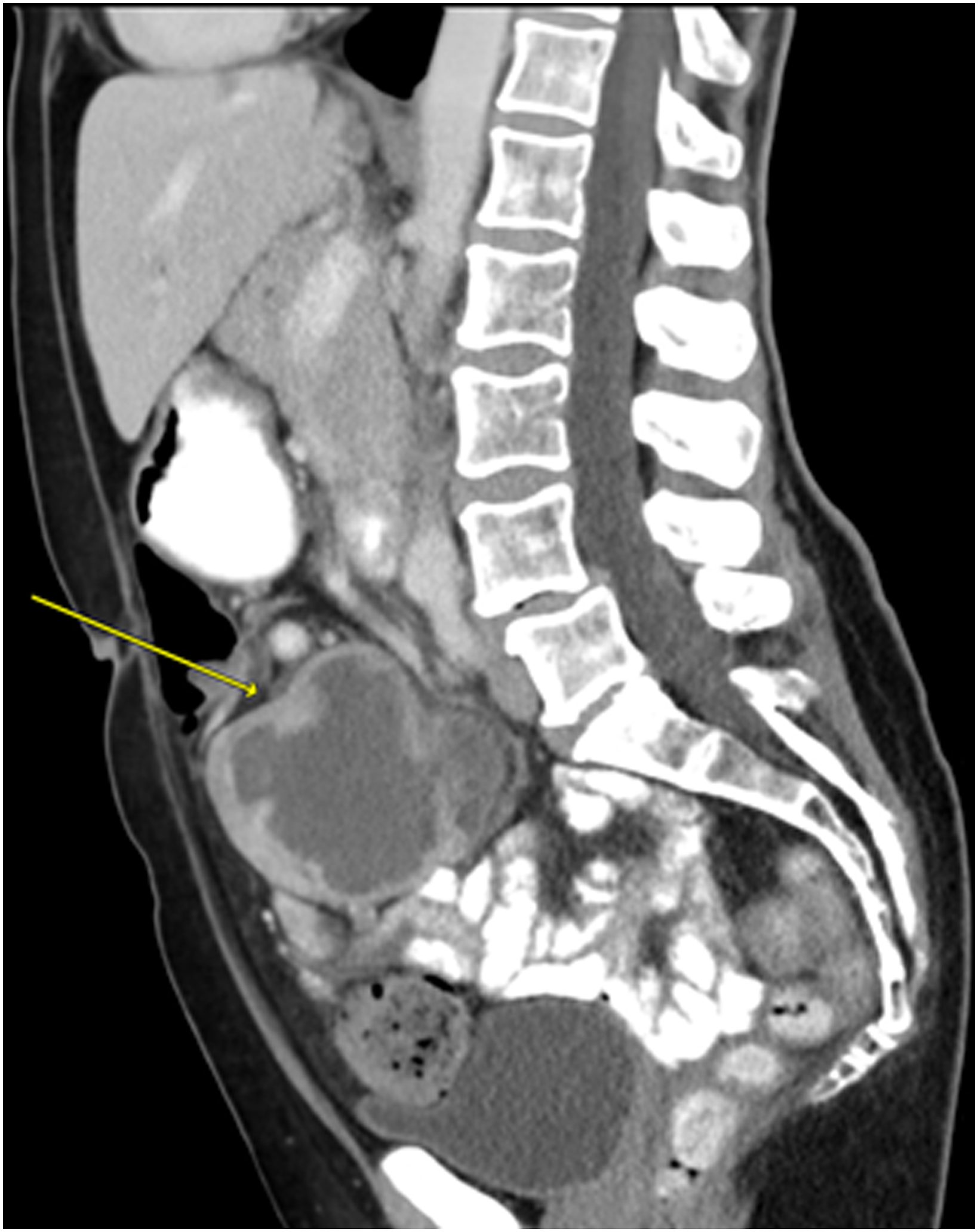
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.