
Abstract
Scientific facts are an essential element of modern life. Yet, despite an abundance of illuminating work in science and technology studies (STS), we lack a rigorous sociological framework for explaining how scientific facts emerge, gain traction, and then change over time. This article fills that gap. Drawing on Fleck ([1935] 1981), Haydu (1998), and a wealth of STS scholarship, I develop a comparative-historical approach called “reiterated fact-making” that analyzes scientific facts according to (1) the prevailing conditions of possibility; (2) the networks of expertise, social mobilization, and power built up around them; and (3) the epistemic and material path dependencies that accrue over the course of their careers. To demonstrate the utility of this framework, I draw on a mixed-methods study and explain the uneven histories of three genetic variants (XYY, Fragile X, and the 22q11.2 microdeletion) that have been used to delineate and diagnose new medical conditions associated with neurodevelopmental differences for the past several decades—taking on very different scientific, clinical, and social meanings as facts in the process. Reiterated fact-making helps us combine comparative-historical sociology and STS to explain how scientific facts can combine deep continuity and radical transformation as they are enrolled in shifting fields of research and practice.
Keywords
Scientific facts are an essential element of modern life. They shape the way we understand and act on the world around us, other people, and even ourselves (e.g., Benjamin 2016; Foucault 1990, 2010; Fourcade 2016; James [1907] 2004; Mead 1967; Shapin 2019; Weber 1946). Given that you are reading this, you probably agree that facts should play a central role in all sorts of decision-making, from the global response to climate crisis to your own health. At the same time, scientific facts are constantly in flux. I do not mean just this or that measurement or statistic. 1 Instead, I follow Ludwik Fleck’s ([1935] 1981) pioneering work on facts and examine the very objects and categories of scientific knowledge production—think gravity, the economy, autism, or genes (all subjects of notable works in the sociology of science). Our often-contested knowledge about these fundamental scientific facts can change incrementally or radically, while many are eventually relegated to the dustbin of history. What we do with them is perhaps even less stable. Indeed, the back and forth between knowledge and practice—the dialectic between what we think we know about a scientific fact and what is done with or about it—is a foundational issue in the social studies of science (e.g., Bowker and Star 2000; Epstein 1996; Fleck [1935] 1981; Frickel and Moore 2006; Hacking 1999; Howlett and Morgan 2010; Jasanoff 2004; Latour 1999; Mol 2002; Shapin and Schaffer 2011).
Yet, despite an abundance of illuminating work in science and technology studies (STS), we lack a sociological framework for explaining the many-varied histories of scientific facts. This article attempts to fill that gap. How, I ask, can we explain the way a fact is established and then goes on to morph and change as an object of knowledge and practice? How does our understanding of a fact at a particular moment—what it is called, what it is, and what is to be done about it—create durable legacies that enable and constrain change down the road? What can a comparative-historical analysis of a scientific fact tell us about the changing social worlds and landscapes for knowledge production it inhabits?
This article proposes an explanatory framework called reiterated fact-making. I follow Haydu (1998) in “making use of the past” to ground a comparative-historical approach to scientific facts. The great virtue of Haydu’s “reiterated problem solving” approach is that it allows us to compare how ongoing issues of social contestation, or problems, are resolved in different historical periods. In this way, we can explain contrasting outcomes without losing sight of the way prior settlements shape the scope for action later on. But what if, instead of social problems, we study the way objects of knowledge, or facts, change over time? For reasons I turn to later, adapting reiterated problem solving to the study of scientific facts requires deep revision. Hence, I turn to the work of Ludwik Fleck. Back in 1935, Fleck showed how facts are not simply given to us by nature, but forged by historically embedded groups of experts and stakeholders and subject to profound change over time. Fleck forcefully argued that facts undergo a process of development that includes everything from experimental innovation to the cultural biases of a scientific community and the ability to resonate with lay publics. Fleck provides a versatile framework for analyzing the way facts move between different fields and publics—variously becoming objects of intense contestation or taken-for-granted reality—and how they change and gain power in the process.
Reiterated fact-making helps us explain how scientific facts emerge, achieve some degree of traction or stability, and then change over time as they are enrolled in new fields of research and practice. Combining Fleck and Haydu—alongside a wide range of insights from the sociology of science and STS—it provides a comparative-historical framework for analyzing (1) the prevailing conditions of possibility at different time periods; (2) the shifting networks of expertise, social mobilization, and power organized around a fact; and (3) the material and epistemic path dependencies created along the way. Together, these factors shape what a fact is, what it means, and how it can be used.
To demonstrate the analytic utility of reiterated fact-making, I use it to trace the varied histories of genetic mutations or variants 2 that have been used to carve out new medical conditions associated with neurodevelopmental differences and congenital anomalies in the United States since 1959. To be sure, humans have always been affected by genetic variants. However, that is just the long pre-history to a comparative-historical analysis of these facts. It was not until the late-1950s and early-1960s that human chromosomal abnormalities or mutations began to be observed in cytogenetics labs and reported in journal articles. In other words, even the very first genetic variants to be discovered have been scientific facts for only the past 60 years or so.
Over those past six decades, genetic variants have married deep continuity and radical transformation. Chromosomal abnormalities like trisomy X or 13, XYY, 5p minus, and 18p deletion have been constants in the literature since the early 1960s, and they have been carefully maintained as consistent objects across several generations of genetic testing technologies. Hundreds more genetic variants joined them as well-established facts in the years that followed. Yet, what it means to have one of these variants depends on a person’s place, race/ethnicity, social class, and many other dimensions of inequality. It has also changed in profound ways over time. The expectations for development, recommended surveillance and treatment regimes, communities, and social identities that accompany the discovery that a person has a given genetic variant—even the way we understand their pathophysiology—can all shift dramatically. The very phenotypes associated with them are often markedly different as well. These are the sorts of transformations and continuities reiterated fact-making can help explain.
This article has five main parts. First, I sketch the broad contours of our case study. Next, I provide an overview of some of the most cognate work in the sociology of science and knowledge, STS, and the history of science. Then, I outline the rationale and core features of reiterated fact-making as a comparative-historical framework and lay out its three explanatory dimensions. In the main empirical section, I discuss the findings from a mixed-methods research project and use reiterated fact-making to explain the uneven history of three genetic variants as scientific and social facts: XYY, Fragile X, and the 22q11.2 microdeletion. Finally, by way of conclusion, I discuss the broader utility of reiterated fact-making as a comparative-historical tool for the sociology of science and knowledge.
Genetic Variants and Neurodevelopmental Differences in Humans
In 1959, researchers studying chromosomes under the microscope reported differences in what we would now call human genomes for the very first time. It started with a 1959 paper in Nature reporting an XXY sex chromosome complement in a Klinefelter syndrome patient (Jacobs and Strong 1959) and was catapulted into international fame later that year with the discovery by Lejeune, Gautier, and Turpin (1959) that trisomy (or three copies rather than the usual two) of what is now called the 21st chromosome appeared to be the cause of Mongolian Idiocy, later renamed Down syndrome. But just months later, researchers found a new chromosome abnormality that did not seem to line up with an existing condition: so-called “Super Females” (Jacobs et al. 1959) with three X chromosomes, or what we now call trisomy X, most of whom are mildly or barely affected. Over the following decades, human geneticists identified hundreds of chromosome abnormalities and genetic mutations (hereafter, usually “variants”) (de Chadarevian 2020; Harper 2006). Some appeared to cause consistently serious congenital anomalies or developmental disabilities, while others were highly variable or mild. A small minority of these variants seemed to explain established disorders. Some variants forced researchers to reconsider the boundaries of conditions like cystic fibrosis and Huntington’s disease, or to lump old diagnoses together and split others apart (see, e.g., McKusick 1969; Miller et al. 2005). Many other genetic variants, however, led to “genomic designation” (Navon 2011, 2019; see also Ledbetter 2008): they were used to carve out entirely new conditions associated with neurodevelopmental differences, congenital anomalies, and medical issues—from the XYY and 5p minus syndromes in the early 1960s to the NGLY1 and ADCY5 single-gene disorders of the past few years.
In this article, I focus on this latter group of genetic variants associated with genomically designated conditions. Why? First, these sorts of variants have unusually long and consistent histories as facts in the biomedical literature. 3 Second, being tied to a genetic variant gives disease categories unusually clear and bounded histories as scientific facts, facilitating comparative-historical casing. We can trace those facts back to the moment the variant was first reported in a journal or database and follow its history from there. Despite the many challenges of identifying and characterizing a new variant in the lab (see de Chadarevian 2020; Hogan 2016; Lindee 2008; Rabeharisoa and Bourret 2009; Timmermans 2017), not to mention the sociotechnical work often required to maintain continuity across different generations of genetic testing platforms, once they are established and “black-boxed,” a variant can provide unusually clear necessary and sufficient conditions for diagnosis. To be sure, historians and STS scholars have written incredibly illuminating analyses of other genetic disorders, including sickle cell (Wailoo 2001), Tay-Sachs (Lindee 2008; Wailoo and Pemberton 2006), and cystic fibrosis (Hedgecoe 2003; Kerr 2005). Still, the specificity that comes with genomic designation allows us to follow the variants as facts as they travel through different fields and morph over time, making them especially useful for a discussion of a new comparative-historical framework.
When a genetic variant is first reported in a journal or database, it has become a scientific fact—albeit sometimes a tentative, partial, or contested one. But even when a novel variant is reported in a top journal like The Lancet or the New England Journal of Medicine, as many of the first chromosomal abnormalities were, it still has a long way to go before it becomes the kind of fact that truly matters to clinicians, patients, and families. Discovering a variant could certainly make a geneticist’s career. But for decades, these variants simply did not matter all that much outside of academic human genetics research. Throughout the 1960s and 1970s, there is no record of these variants leading to major changes in patient care, never mind the rich social lives that would follow for some. A genomically designated condition could explain the presence of neurodevelopmental issues and congenital anomalies, and by the mid-1970s a growing number of variants could be used to inform selective abortion, but otherwise they had very little impact in applied fields (Harper 2006; Lindee 2008).
Today, by contrast, finding one of these genetic variants can profoundly shape patients’ lives. A genetic diagnosis can become far more than a mere explanation—it can inform treatment, prospect horizons, community, activism, and identity (Hacking 2006; Lindee 2008; Panofsky 2011; Rabeharisoa, Moreira, and Akrich 2014). In some cases, one finds detailed management guidelines and specialist clinics spanning a panoply of medical disciplines. Knowing that a child has a particular variant can even inform practice in fields like psychology and special education (Finucane, Haas-Givler, and Simon 2003; Reilly 2012). A range of scientific fields have turned their attention to genetic conditions and variants, forever changing how we understand everything from their pathophysiology to the experiences of the people who have them. These variants have been leveraged as biological models by researchers seeking to unlock the genetic basis of more common conditions like autism, schizophrenia, and obesity, and even our capacities for sociality or aggression (e.g., Sahin and Sur 2015). Social movements have organized around some variants, with support groups, research repositories, and registered foundations dedicated to the cause. A growing number of variants have been taken up by popular media and filmmakers, activists and politicians, research institutes, and pharmaceutical companies. Along the way, our rendering of these variants’ very phenotypes have evolved from thin lists of a few associated congenital anomalies and cognitive delays to richly detailed portraits of different medical issues, neurodevelopmental differences, and fine-grained behavioral idiosyncrasies.
Most genetic variants, however, have not undergone such a transformation. That is why we need a comparative-historical account. We need to explain how some variants go from objects of specialist academic research and little more, to facts that can guide practice across a wide range of fields, animate a mini-social movement, and transform the meaning of illness, disability, and difference.
Intellectual Background and Literature Review
As a wealth of scholarship in STS and the history of science makes abundantly clear, scientific facts are often caught up in processes of social change. Our collective responses to a scientific fact can lead not only to important changes on the ground, but also fundamental revisions to the fact itself. As Bachelard put it (cited in Latour 1999:127) “un fait est fait”—a fact is fabricated. Indeed, the Latin root of fact, facere, meaning “to make, do, or accomplish,” (see Knorr-Cetina 2005:225) lived on in the original English use of fact: a thing that is done, performed, or achieved. That does not mean facts are somehow fake or a mere “social construction” (Hacking 1999). But it does signal that scientific facts are made up of different elements, combined in knowledge production and practice, and always subject to perturbation, reconfiguration, rejection, or even being a fundamentally different or “multiple” thing across sites (Daston 1994; Eyal 2013; Latour 1999; Mol 2002; Rheinberger 1997).
Making matters even more complicated, scientific facts are often surprisingly contingent and unstable. For one thing, they often have precarious origins. Modern STS scholarship famously showed how objects of knowledge emerge in strongly context- and equipment-dependent sites, like laboratories, and get codified as facts through the careful inscription practices and political positionings of scientific entrepreneurs (for influential accounts, see Knorr-Cetina 2005; Latour 1988, 1993; Rheinberger 1997). As Latour and Woolgar (1986:105) put it, these kinds of studies reveal “the precise time and place in the process of fact construction when a statement became transformed into a fact and hence freed from the circumstances of its production.”
For another, scientific facts can change beyond recognition or lose their status as facts altogether. Indeed, the idea that facts and even entire scientific fields can undergo radical change or crumble away lies at the very heart of the social studies of science and knowledge (Bachelard [1938] 2002; Fleck [1935] 1981; Foucault [1969] 2002, 1973; Kuhn 1996). Some objects of knowledge are treated as facts for a time, but then fall out of favor—phlogiston, miasma, or spontaneous generation to take a few notable examples. Others, like the “gene,” undergo such a fundamental reformulation that we may struggle to treat them as consistent facts across fields or historical periods (Keller 2002; Rheinberger and Müller-Wille 2018). A volume on the Biographies of Scientific Objects explored what Daston (2000:1) calls “applied metaphysics,” or work that “studies the dynamic world of what emerges and disappears from the horizon of working scientists.” More broadly, Daston and others have created a subfield of “historical epistemology” that examines the uneven status of scientific concepts and objects—or even what constitutes facticity itself—over time (e.g., Daston 1991, 1995; Davidson 2004; Poovey 1998; Rheinberger 2010; see also Hacking 2004 on “historical ontology”). I am deeply indebted to Daston and her fellow travelers.
However, I depart from these prominent threads in the history of science/STS in two ways. First, I do not confine my analysis to the horizons of working scientists, their concepts, or their experimental apparatuses. Second, I am equally interested in the less spectacular, but often more sociologically important segment of a fact’s scientific career that is characterized by a degree of durability: when a scientific fact maintains its coherence even as it is caught up in processes of transformation. Scientific facts may be even more likely to affect our lives when they are “black-boxed” (Latour 1988) and taken for granted qua facts. That is why I study the way scientific facts emerge and then dynamically interact with the world around them over the course of years and decades. After all, some of the most important scientific facts do not just fade away—they change with the times, maintain their status as objects of knowledge and practice, and continue to play a key role in many different arenas of social action and power.
To be sure, creating and maintaining a durable scientific fact is no small feat. A scientific fact can never be just the same as the thing it is designed to capture—something ostensibly out there in the world that is independent of our knowledge about it. Whether and how those “noumena” actually exist is a problem our colleagues in philosophy have wrestled with for centuries, but it need not detain us here. Suffice it to say there is no final answer, no version of a scientific fact that will prove immutable and beyond reproach. Scientists do not paint on a blank canvas or divine the world on a whim. But science also has to intervene in the world in order to represent it, and objects of knowledge often push back against or “overflow” our understanding of them—a key source of scientific innovation we will encounter later on (Callon 1998; Hacking 1983; Latour 2007; Rheinberger 1997).
Consider our case study. Pathogenic variants do not simply reveal themselves as distinct, fully formed facts the moment researchers deploy a suitable genetics technology (de Chadarevian 2020; Harper 2006; Hogan 2016; Latimer 2013; Rabeharisoa and Bourret 2009; Timmermans 2017). Testing for a variant only becomes a binary question about a person’s genome after a great deal of work. These variants start out as what Rheinberger (1997) calls “epistemic things”—unpredictable objects with no clear referent outside of the “experimental system” in which they come into view, be it karyotyping in the 1960s or genomic sequencing today. Over time though, they are often stabilized or “black-boxed” and turn into what Rheinberger (1997:30) calls “technical things” (see also Latour 1988 on black-boxing) that can be studied across different testing platforms and research programs. In this way, a variant may cease to be an epistemic thing in the world of genetic testing. As Fleck teaches us, it is precisely this movement out of the lab and into new fields and publics that allows a finding to shed its tentative status and become an apodictic scientific fact. But that is merely the end of the beginning. Once variants are rendered provisionally stable as genetic test results, they can begin new and disruptive careers across an array of scientific and social fields. This is the kind of trajectory we can analyze with reiterated fact-making.
More broadly, reiterated fact-making can help sociologists generate findings akin to some of the most incisive work in the history of medicine. Wailoo’s magisterial book charting the history of sickle cell disease and black suffering in Memphis, Dying in the City of the Blues (2001), is one especially notable example. So too is Rosenberg’s influential book, The Cholera Years ([1962] 2009), which incisively compares three mid-nineteenth-century cholera outbreaks in the United States (for a sociological take, see Whooley 2013). Important works in the history and philosophy of science have examined facts and concepts across eras, from Canguilhem’s ([1943] 1991) work on pathology and Jacob’s ([1970] 1993) book on the structure of life to Foucault’s ([1961] 2009, 1990) books on madness and sexuality. More recent historical work in STS has followed in their footsteps—Keating and Cambrosio (2006) on immunophenotypes, Pickering (1999) on quarks, Valverde (1998) on alcoholism, Fullwiley (2011) on the gene involved in sickle cell, Landecker (2010) on cells, Franklin (2007) on sheep, and Davidson (2004) on sexuality, to cite a few notable examples. Sociologists of knowledge have also interrogated different iterations of key facts, like “the economy” (Mitchell 2008), HIV/AIDS (Epstein 1996), and even “nature” (Fourcade 2011).
Finally, when it comes to categories of human difference, scientific facts are further subject to what Hacking (1998, 2007) calls “looping” processes. When experts classify people with, say, a mental illness, the expectations and interventions that come with the diagnosis may change them. However, those changes will often defy expert predictions—via unintended consequences, resistance, or what have you—leading experts to revise the classification. The category is then applied to people anew, changing them in new ways, and setting off a potentially endless series of loops between expert knowledge, practice, and the people who are subject to classification. That is how “kinds of people” become “moving targets,” or inherently dynamic scientific facts. As we will see, using genetic variants or other biomarkers to classify people may change looping dynamics, but it certainly does not foreclose them (Navon 2023).
I have drawn invaluable lessons from these and many other works in the history and social studies of science. To be clear, my point is not that the field has ignored the histories of scientific facts. Far from it. However, we lack a comparative-historical framework for analyzing how facts not only emerge but then persist amid tumult and change.
Reiterated Fact-Making
Reiterated fact-making provides a comparative-historical framework for explaining the historical transformation of scientific facts, even when their transformation is underwritten by a deep continuity. It also helps us generate sociological insight from the longue durée of facts’ scientific careers and social lives. Like Fleck, I argue we can learn an enormous amount by tracing “the genesis and development of a scientific fact” (to quote the title of his 1935 masterpiece). This section outlines the twin theoretical pillars of reiterated fact-making before delving into its three axes for explaining the histories of scientific facts.
To be clear, I am not using “fact” to denote the details or pieces of information about something—the often-contested statistics, measures, predictions, and “stylized facts” (see Hirschman 2016) that are so central to “regulatory” or policy-oriented science (cf. Eyal 2019; Oreskes and Conway 2010). Instead, I follow Fleck’s lead and focus on facts in the sense of things that are taken to be real and about which various things are held to be true: “facts” in the sense of “something that has actual existence,” to quote the first definition provided by Merriam Webster. The details of these facts, and what is to be done about them, are often hotly contested in policy circles and many other arenas (including the burgeoning worlds of misinformation and “alternative facts”), even when no one questions their status as scientific facts per se.
Our starting point is Ludwik Fleck ([1935] 1981). Although Fleck is best known for his notion of the “thought styles” that enable and constrain knowledge production, 4 the bulk of his analytic work lay elsewhere. He was doggedly focused on the centuries-long development of a very particular scientific fact: syphilis and its eventual delineation according to the Wassermann reaction. Having worked in the venereal section of a large hospital, Fleck ([1935] 1981:22) was convinced “it would never occur even to a modern researcher, equipped with a complete intellectual and material armory” to isolate “syphilis” from the universe of disease they encountered. He showed how syphilis had changed dramatically over the years as disciplinary alignments, background assumptions, and cultural imperatives shifted and churned. It took “cooperative research, supported by popular knowledge and continuing over several generations” for syphilis to coalesce as fact (Fleck [1935] 1981:22). Nor, Fleck noted, would the Wassermann reaction likely be the final say on “syphilis”—a prescient observation.
Fleck was perhaps the first to truly grapple with the dynamic relationship between primary research on a scientific fact, applied fields, and popular knowledge. He was fascinated by the relationship between the highly specialist “esoteric” researchers who develop preliminary knowledge about a fact and the “exoteric” or external fields and publics who variously engage, stabilize, and transform it (Fleck [1935] 1981:113). Fleck discussed the path an object of knowledge must take, from a vanguard of esoteric researchers with their “journal science” to the “vademecum science” of reviews and encyclopedias, and eventually to the kind of “popular knowledge” that stabilizes a fact and “reacts in turn upon the expert” (p. 113). He drew on the example of a bacteriological exam whose uncertainty and complexity can only be resolved once a laboratory technician can provide a concise lab report to a general practitioner, who in turn can provide a clear diagnosis to a patient (e.g., “your child has diphtheria”) (p. 115). Only then, Fleck ([1935] 1981:125) argued, a “fact becomes incarnated as an immediately perceptible object of reality.” Fleck ([1935] 1981:115, emphasis in original) called this “the general epistemological significance of popular science”: “Certainty, simplicity, vividness originate in popular knowledge. That is where the expert obtains his faith in this triad as the ideal of knowledge.” Fleck was far ahead of his time. He showed how facts are not made by scientists alone, but by a whole range of actors—all living in particular places and historical moments that shape their notion of what is knowable and possible. If we want to understand a scientific fact, its history, and its power, we therefore need to examine how that fact not only emerged, but also how it was fundamentally transformed as it reverberated between different fields of experts, stakeholders, and publics. In sum, Fleck provides the theoretical foundation for a sociological approach to scientific facts.
But what would a comparative-historical approach to scientific facts look like? In a way, I am harkening back to a classic form of comparative-historical sociology that aims to explain outcomes through qualitative analyses of multiple cases. But to analyze something like genetic variants over time, the conventional comparative logic is clearly not viable. For one thing, studying dependent variables would merely scratch the surface of our sociological interest in scientific facts. For another, scientific facts do not lend themselves to the comparative logic of the Millian method (Mill [1843] 2002), or any variation of it (for the most influential rendering in sociology, see Skocpol 1979). The comparative elements within this article’s cases, for example, are anything but independent from or equivalent to one another, rendering the Millian method a non-starter (Sewell 1996): researchers and other actors very deliberately draw on important work on other genetic variants, not to mention prior iterations of their own variant of interest; meanwhile, the landscapes for studying and acting on pathogenic variants vary widely across time and cases.
To overcome these obstacles, I build on Haydu’s (1998, see also 2010) “reiterated problem solving” framework. Like Haydu, I want to deploy both narrative and path dependency approaches, allowing us to embrace historically specific conditions and contingencies as part of a causal analysis. This allows for a comparative analysis of different historical settings, a narrative explanation of different outcomes (see Calhoun 1998; Somers 1994), and careful attention to the way settlements in one period both constrain and enable actors working to solve a homologous problem later on. Reiterative problem solving therefore holds great promise for the diachronic comparison of scientific facts.
However, I cannot truly claim to be “connecting events between periods through sequences of problem solving” (Haydu 1998:349). Why? Haydu (1998:355) insists that in delimiting reiterated problems “one criterion must be the social actors’ own understandings . . . there must be some correspondence between the observer’s conception of a recurring problem and the social actors’ experiences of confronting common obstacles and devising ways to surmount them.” Unlike Haydu, we cannot use a fixed set of actors confronting a similar “problem” as our casing. Despite important continuities over the half-century or so covered in this article, we cannot assume human geneticists always had the same goals vis-à-vis pathogenic variants. Nor should we confine ourselves to the field of human genetics. We do not want to treat the work of patient advocates, genetic counselors, and other key groups as “exogenous” (as Haydu [1998:364] frames state intervention during the formulation of a new regime of labor relations in the 1930s). Human geneticists in the 1960s and a patient advocate today would not “recognize a common dilemma” or a sense of what was possible (see Haydu 1998:361), even if their interest lay in the same genetic variant. Instead, new sets of actors entering the fray and fundamentally reframing notions of what is possible, that is, the salient “problems” of knowledge and practice, is often central to a scientific fact’s career.
Our casing instead draws on deep continuities in the facts at hand, not a constant set of actors and interests. A comparative-historical framework that focuses on a scientific fact can therefore use shifts in who is working with them and what they are trying to achieve (i.e., the problems they are working to solve) to explain change over time. This allows us to examine how different configurations of actors variously construct a scientific fact across historical contexts. A scientific fact may simultaneously function as a social problem, but whether and how that works can shift in profound ways. So, if reiterated problem solving “puts social actors at center stage” (Haydu 1998:357), reiterated fact-making casts facts themselves as protagonists in our saga. It allows for the possibility that new actors will not only enter in Act II or III, but drive major plot twists as well.
Reiterated fact-making therefore provides a framework for tracing facts across different places and time periods, the changing networks built up around them, and the divergent forms of knowledge and practice they give rise to. This allows for a comparative analysis of, as Callon (1995:54) puts it, the way a fact’s “identity depends on the state of the network and the translations under way, that is on the history in which they are participating,” bringing the causal explanation offered by historical sociology (e.g., Ermakoff 2019) to topics typically addressed solely in STS. That means following Fleck’s example and analyzing everything from the shifting sociocultural conditions that operate in the background through to the fine-grained ways facts move between fields. Crucially, it helps us grasp how the facts themselves may be simultaneously transformed and held stable.
In short, reiterated fact-making provides a framework for explaining how scientific facts emerge and are rendered more or less stable even as they change over time in concert with the various actors, objects, and fields they encounter. That means we need to look at three factors, summarized in Figure 1. The rest of this section discusses how each of these factors can help us explain the way scientific facts are made and remade over time.

The Three Explanatory Elements of Reiterated Fact-Making
Conditions of Possibility
What must change in the world to make a particular scientific fact thinkable and meaningful? What sets the stage for it to emerge as an important object of practice? How do “circumstances existing already, given and transmitted from the past” (Marx 1852) enable and constrain the way we make knowledge in the present? These questions force us to confront the prevailing conditions of possibility for a scientific fact. These are not quite what comparative scholars have called the “necessary causes” for a given historical outcome. Instead, I want to get at the exogenous historical developments and non-trivial background conditions that make it possible for a scientific fact to be discovered, understood, or used in a certain way.
The concept of conditions of possibility originated with Kant and his transcendentalist attempts—especially in the Critique of Pure Reason ([1781] 2003)—to overcome Hume’s philosophical skepticism. Kant famously argued, for example, that we cannot conceive of a three-dimensional object that does not exist in space or imagine experiencing the world without causality. Thus, he concluded, we can sketch certain “synthetic a priori . . . conditions of the possibility of the objects of experience” (A158/B197). Centuries later, Foucault ([1969] 2002) provided a sociological take on the concept. Foucault argued that any statement, fact, category, theory, or what have you is only intelligible within the broader frameworks that govern thought and knowledge production at a given time (see also Daston 1995; Hacking 1999; Poovey 1998). In other words, our modes of understanding and knowing are historically specific, be it the idea that diseases are traceable to organic lesions (Foucault 1973) or the modern concept of sexuality itself (Foucault 1990). In later work, Foucault (1977a:194–95, 2010:19–20) expanded his focus to include a host of other factors—political, cultural, institutional, and so on—that together form an apparatus or dispositif that makes any given complex of knowledge and power possible.
Some conditions of possibility are so obvious they teeter perilously close to what comparative-historical scholars have called “trivial necessary causes” (see Mahoney 2004:82–83), like humans as a cause of revolutions or gravity for a military victory. Other factors, by contrast, need to be carefully unpacked in a sociological explanation of a fact. This is an interpretive process, but one that can be applied quite systematically. The key here is to home in on pertinent exogenous factors and then unpack how they make a particular rendering of a fact conceivable or doable, that is, possible. The analyst therefore needs to immerse themselves in the history of a scientific fact and use inferential and counterfactual reasoning to separate the explanatory wheat from the trivial chaff (for an illuminating discussion, see Abend 2020:84–85). That is why our empirical case study will grapple with developments in genetic testing and data dissemination, institutionalization and de-institutionalization, and the rise of modern patient advocacy as key conditions of possibility driving change over time, but not the evolutionary synthesis, representative democracy, the dominance of allopathic medicine, or any number of other things that could have mattered in theory, but instead serve as constant background conditions for a study focused on genetic difference in the United States between 1959 and the present. In this way, we can identify shifting historical conditions and “epistemic cultures” (Knorr Cetina 2009) that are exogenous to a scientific fact, but nevertheless essential to a narrative explanation of its emergence and development.
Networks of Expertise, Social Mobilization, and Power
It takes actors to delineate and establish a scientific fact, never mind turn it into an important object of power and practice. So, while conditions of possibility operate in the background—making a fact thinkable and potentially actionable almost inadvertently—networks of expertise, social mobilization, and power are developed around a fact: to establish it, study it, promote it, commercialize it, prevent it, treat it, mitigate its impact, or what have you. These networks are directly engaged with a scientific fact, from discovery and refinement through to application, reaction, resistance, and even refutation. Our cases help crystalize this distinction. Despite increasingly favorable conditions of possibility, most pathogenic variants are still not mobilized to any great degree. Clearly, conditions of possibility radically underdetermine scientific facts. To riff off Kant, space makes three-dimensional objects possible, but actors can craft and use the things that inhabit that space in all manner of ways.
Contemporary STS, and especially actor-network theory, shows us how facts are assembled through the interventions of heterogeneous networks of experts, social entrepreneurs, and nonhuman “actants” (Callon 1995; Latour 1988; Law 1992). This article draws extensively on that literature. However, rather than focus solely on experimental laboratory research (cf. Latour and Woolgar 1986; Rheinberger 1997) or how a central actor “translates” the interests of the others via some sort of new fact (cf. Callon 1986; Latour 1993; Star and Griesemer 1989), reiterated fact-making shows how scientific facts themselves change even after they are firmly established, as the networks organized around them shift over time. This approach is therefore more akin to Eyal’s (2013) focus on “networks of expertise,” which extends the sociology of professions by following scientific categories, technologies, and so on well beyond the realm of credentialed experts and formal knowledge (see also de Laet and Mol 2000).
Two key points should be highlighted here. First, the networks built around facts are composed of both humans and non-humans. This includes the objects of knowledge themselves, be it microbes for Latour (1993, 1999) or genetic variants in this article: paradoxically, they are at once dependent on the tools, labs, models, databases, and applied or commercial fields in which they circulate as facts and capable of exerting a degree of “agency” when their unexpected behavior and “overflow” effects (Callon 1998; Latour 1988; Rheinberger 1997) drive discovery and innovation. Moreover, to make sense of a genetic variant or any other scientific fact, you need written and graphical “inscriptions” that can represent scientific observations and then circulate throughout the network, or what Latour (1988:226–37) called “immutable and combinable mobiles.” That is how a fact can travel far beyond the site of basic research—a process Fleck showed can take uncertain esoteric knowledge and turn it into the kind of apodictic fact that has traction across both expert and public fields. In one way or another, virtually everything we know about a scientific fact is mediated by tools, inscriptions, interventions, and what Edwards (2010:17) calls “knowledge infrastructures”: the “robust networks of people, artifacts, and institutions that generate, share, and maintain specific knowledge about the human and natural worlds” (see also Hirschman 2021). Second, when these networks change, so do the facts. This is why Fleck’s insights about the relationship between esoteric, exoteric, and popular fields is so essential: facts develop novel contours and applications—not to mention sites of potential contestation—when they circulate in new scientific disciplines, fields of practice, and publics.
Finally, reiterated fact-making’s focus on these broader networks also helps us interrogate the many intersecting dimensions of inequality at play in an analysis of scientific facts. Different groups of experts (lay or credentialed) have unequal authority to delineate and shape scientific facts, while stakeholders have radically uneven levels of privilege and autonomy when they encounter a fact—making it variously a source of edification, utility, power, interpellation, domination, and so on. So, by examining the uneven and ever-shifting network of human and non-human actors assembled around a scientific fact, we can understand how it emerges and is held stable even as it changes over time and across different settings.
Path Dependencies
Scientific facts are not born anew every time the next cohort of researchers decides to study them. Nor are they renegotiated from scratch every time they intersect with new worlds of practice or culture. Facts are always at risk of being discarded, marginalized, or overhauled, but they also have a durability or “stickiness” thanks to prior investments in knowledge production, material infrastructures, and cultural meaning. We therefore need to pay careful attention to the path dependencies that shape the way we understand and use scientific facts.
Historical sociologists, economists, and political theorists have long drawn on the notion of path dependency to capture the way contingent events in one period can set deterministic processes in motion and lock seemingly underdetermined structures in place (for detailed sociological discussions, see Goldstone 1998; Mahoney 2000). Scholars have used path dependence theory to explain everything from the adoption of technologies and standards like the QWERTY keyboard (David 1985; for other examples, see Arthur 1989), to different post-Communist trajectories after the fall of the Soviet Union (e.g., Róna-Tas 1997), divergent patterns of regional economic development (Martin and Sunley 2006), and a variety of major social policies (e.g., Peters, Pierre, and King 2005). In short, path dependency has been a key tool for incorporating contingency into the narrative explanation of small-n historical developments.
But how should we think about path dependence when it comes to a scientific fact? To understand the history of a fact we need to account for two overlapping sorts of path dependency. First, there are the epistemic path dependencies that shape the way facts are understood. Prior findings, definitions, measures, and standards always loom large. So do prior renderings of a fact. They can be overturned, but never at zero cost. Indeed, they are often maintained at great expense in the name of historical continuity and comparison. If a new rendering of a fact does not conform to the prevailing scientific orthodoxy, it will likely be “subsumed” and thereby relegated (Decoteau and Daniel 2020). Second, there are the material path dependencies that create scope conditions for understanding and acting on facts. This includes the tools we use to detect and measure a fact; the interventions, infrastructures, and devices developed in response; the repositories and databases researchers use; and so on. As we will see for genetic variants in humans, contingent developments can reverberate for decades, shaping the networks of knowledge production and practice that form around scientific facts through many kinds of epistemic and material path dependencies. In this way, as we see in Figure 2, exogenous conditions of possibility and path dependencies from the past combine to shape the current state of network formation around a fact, and with it the fact’s status as an object of knowledge and practice.
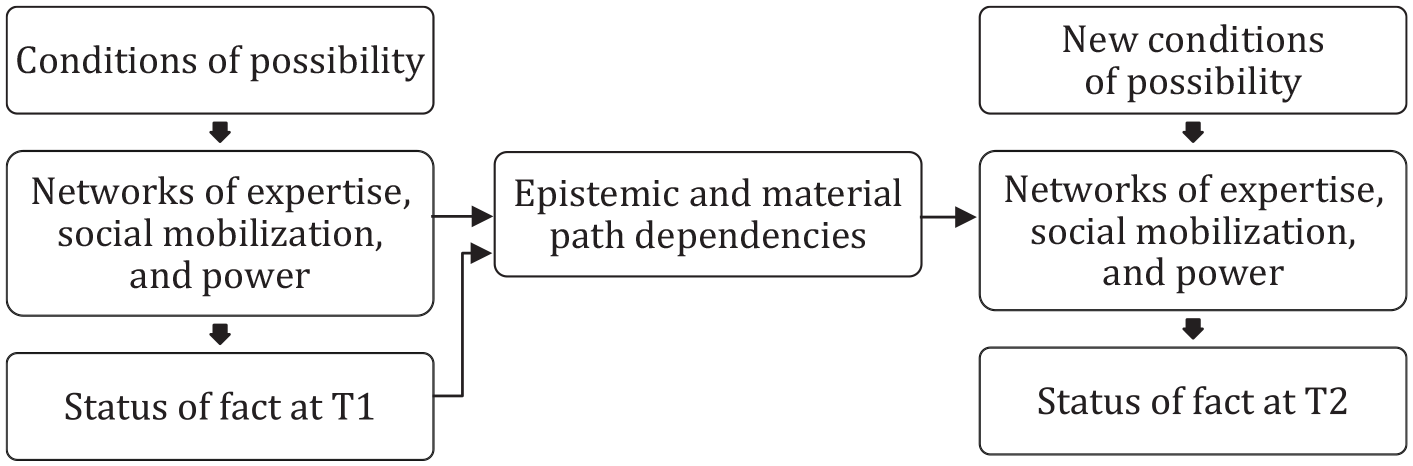
How Reiterated Fact-Making Explains Continuity and Change in a Scientific Fact across Time Periods
This is a flexible model. Periodization is a matter of interpretive discretion, depending on what it is about a fact that one is trying to explain and the pivotal developments encountered during research. The model might be used to explain the transition from T1 to T2, as in Figure 2, to continue to T3 and on to Tn, or to take a narrative approach that describes a more continuous process of transformation through to the present. The model can also accommodate more or less radical change in the status of a fact over time. Some facts may be tentative or contested at T1 but then widely accepted or “apodictic” by T2. Others may lose their status as scientific facts altogether, like miasma, phlogiston, aether, and their fellow occupants in the history of science graveyard. In many other instances, including this article’s case studies, facts are enrolled in new fields, put to new uses, and attributed different or contested meanings over time. Across a range of outcomes, the model can be used to identify the key background conditions, actors, innovations, events, and serendipities—and to uncover the black-boxed conflicts, interests, assumptions, and mobilizations—that drive change. Whatever the approach and whatever the outcome, reiterated fact-making provides a parsimonious model for explaining the career of a scientific fact.
Making and Remaking Genetic Variants
How can reiterated fact-making help us explain the emergence and development of pathogenic variants as scientific facts? This section draws on a major research project that used reiterated fact-making to analyze the 60-year history of genomic designation as a way of delineating and diagnosing human disease and difference (Navon 2019). The focus is mainly on the United States, although other Western countries featured prominently in the study’s research and findings. Whereas that broader project used reiterated fact-making mostly implicitly across a wide range of cases and findings, this article homes in on just a trio of interlinked cases—the XYY, Fragile X, and 22q11.2 deletion syndromes—to theorize and demonstrate its value as an explanatory framework for sociologists of science.
Methods
Research for this project drew on fieldwork, interviews and oral histories, bibliometric analyses, archival research, and a genealogical analysis of the biomedical literature. Fieldwork was conducted between 2010 and 2015 at six major conferences that brought together experts and families dedicated to specific genetic conditions, as well as several virtual conferences, various smaller events, visits to specialist centers, an array of online forums, and a meeting of the U.S. Department of Health and Human Services’ Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children. All told, this allowed for hundreds of hours of participant observation. I also conducted around 40 interviews with experts and parents.
My historical research combined oral history with an extensive discourse analysis of the pertinent biomedical literatures from the late-1950s to the present; I used a genealogical approach (Foucault 1971, 1977b) to trace the histories of these genetic variants as scientific facts. 5 I also used bibliometric analysis of ISI’s Web of Science and archival research at the Wellcome Institute in London, as well as online archives maintained by the NIH, the New York Times, and several of the foundations dedicated to rare genetic conditions. Throughout, this research was supplemented by extensive discourse analyses of publicly available resources from foundations—including factsheets and consensus clinical guidelines, how-to materials for engaging local clinicians, and sources explaining genetic disorders to affected children—as well as materials from social media, healthcare organizations, public agencies, and major print and broadcast media.
Findings: Three Genetic Variants across Three Eras
Perhaps the most obvious condition of possibility worth mentioning for the scientific facts discussed in this article came into view in 1959: the ability to observe abnormalities in human genomes (as they came to be called). As we saw, the newfound technical capacity to see, count, and label chromosomes, shortly followed by “banding” techniques that could identify specific segments of chromosomes, gave us the first true genetic testing technology. Cytogenetics, as the subfield is known, made it possible for researchers to discover dozens of chromosome anomalies and report them using a standardized typology. This gave genetics its “morbid anatomy” (McKusick 1982) as an aspiring medical subfield, and new variants and syndromes quickly flooded the literature (de Chadarevian 2020; Harper 2006; Lindee 2008).
But a genetic variant’s discovery is just the beginning of its career as a scientific fact. Back in the 1960s and 1970s, other prevailing conditions of possibility virtually guaranteed that most of these newly discovered variants and genomically designated conditions would remain very thin as scientific facts—with little further research, detail, or impact beyond the academy. Cultural and institutional factors need to be reckoned with here. The overwhelming majority of people identified with genetic variants in the 1960s and 1970s were confined to institutions and asylums (de Chadarevian 2020; Lindee 2008). These scientific facts did emerge from a preexisting social problem: populations who had been subject to eugenic confinement (and worse). As Canguilhem ([1943] 1991) noted long ago, scientific medicine usually needs a pathological state to establish biological fact.
Institutionalization made it at once easy for geneticists to locate (and extract samples from) people with the tell-tale signs of genetic abnormality and virtually impossible to use a genetic diagnosis to guide (postnatal) clinical practice or care. Even as the first wave of chromosomal abnormalities was extensively reported in top journals like the Lancet and the New England Journal of Medicine, few doctors could order a cytogenetic test (if the thought even occurred to them), nor did they have the resources to interpret a positive result. Beginning in the late-1960s, and especially after the legalization of abortion in 1973, these variants could have a profound impact on family planning when they were discovered via amniocentesis (Rapp 1999). The limited postnatal cytogenetic testing being conducted, however, was mostly confined to esoteric research studies. Families were mostly not involved. There was no realistic path to build up patient numbers, develop a detailed clinical profile, or pursue meaningful programs of treatment or community formation. Having a genetic variant could clearly matter a great deal to a patient; identifying it usually did not.
The notable exception was XYY: the ~1 in 1,000 males with an extra Y chromosome. XYY similarly emerged as a fact when researchers brought new cytogenetic techniques to bear on captive populations in asylums, prisons, and institutions in the 1960s (Jacobs et al. 1965; Sandberg 1961). Yet, unlike other chromosomal variants during this era, XYY gained widespread traction as a scientific fact due to its articulation with another condition of possibility: widespread moral panic about violent crime. In the 1960s, this social problem combined with institutionalization and other lingering features of the eugenics era to create the conditions for a spectacular episode of scientific and political controversy to unfold around XYY.
What of the networks of expertise, social mobilization, and power organized around XYY? Researchers in human genetics were joined by colleagues in psychiatry and criminology, major institutions like the NIMH and Harvard, and widespread mass media coverage that reached the front page of the New York Times (Lyons 1968) and other popular outlets. Its reported phenotype fit the bill: XYY males were recruited from prisons and asylums and associated with increased stature, acne, mental “subnormality,” and above all a strong disposition toward aggression and crime. XYY was invoked in high-profile murder cases (including a famous serial killer who turned out to be XY) and science/crime fiction. The variant was seen by experts, policymakers, and publics alike as a genetic fact that could speak to a major social problem.
Yet, a series of actors intervened to disrupt this network. Extra Y chromosomes started appearing in males who did not exhibit signs of “mental subnormality” or aggression. Then, when researchers tried to recruit large unbiased samples of children to answer growing questions about ascertainment bias and potential treatments, the network of expertise and sensationalized public uptake around XYY collapsed into failure and delegitimation. Activists concerned about eugenics and children’s rights, like the ACLU and Science for the People, mobilized against XYY research programs, eventually shutting them down amid intense controversy and moral opprobrium (de Chadarevian 2020; Navon 2019; Richardson 2013). Ascertained in medical-penal institutions, mobilized beyond the lab as a tool of social control, and ultimately delegitimated via social activism, the XYY network’s spectacular collapse demonstrates the very limited potential for these sorts of genetic variants to achieve broader traction given the conditions of possibility that prevailed in the 1960s and 1970s. It turned out to be the exception that proved the rule.
The calamitous implosion of the XYY network left enduring path dependencies. For decades, XYY was delegitimated and mostly ignored as a research topic. It remained highly stigmatized, with a wildly biased cognitive-behavioral phenotype that lingered in high school textbooks and outdated reporting. When XYY did reappear in the public domain, it served as an essentializing criminalistic plot device in shows like Law and Order or the “Double Y Chromosome-Work Correctional Facility” in David Fincher’s Alien 3. XYY karyotypes were only likely to be detected as an incidental finding, or on a prenatal test that left expectant parents with a vexed decision about selective abortion. As we will see, XYY eventually reemerged as a radically different fact, but only after decades in scientific purgatory. Meanwhile, most variants languished as thin facts that mattered very little to patients, families, caregivers, or anyone outside of esoteric human genetics in the 1960s, 1970s, and beyond.
How had the prevailing conditions of possibility changed a few decades later? Deinstitutionalization meant that, by the 1980s and 1990s, people with pathogenic variants were likely to live with their families in the local community. Later, an emerging wave of patient advocacy organizations pioneered by HIV/AIDS and breast cancer activists (Epstein 2016) created unprecedented opportunities and social movement “repertoires” (Tilly 1993) for the sort of advocacy that vivifies genetic variants as facts today. The meteoric rise of the autism parent/patient movement (see Eyal et al. 2010) changed the landscape for advocacy around neurodevelopmental difference, and would go on to become a major driver of interest in rare genetic variants (see Navon and Eyal 2014). New genetics technologies were bringing more and more variants into view, even as rates of genetic testing remained extremely low, and means for communication and data-sharing about genetic variants and disorders were still very limited. The conditions were not exactly favorable, but they made it possible for new alliances of experts and families to begin remaking genetic variants as scientific and social facts.
That brings us to the networks of expertise and social mobilization that can turn a pathogenic variant into the sort of fact that can change people’s lives. Perhaps the most transformative network element for a pathogenic variant is the emergence of an effective patient advocacy movement. Just such a movement began to take shape in the early-1980s when a pioneering alliance of Fragile X experts and parent/patient advocates joined forces around a kitchen table in Denver to establish a support group and foundation. “Fragile X mental retardation” as it was known, had been carved out from the broader category of “x-linked mental retardation” (XLMR). In 1969, cytogenetics researchers first observed a “fragile site” on the X chromosome; then, in the early 1990s, a specific CGG trinucleotide repeat expansion on the FMR1 gene became visible with new testing technologies. During this period, Fragile X emerged as a distinct genetic disorder characterized by a craniofacial phenotype, macroorchidism, and what we would now call intellectual disability (Krueger and Bear 2011). Fast forward a couple of decades, and the National Fragile X Foundation (NFXF) had become the central organization in a powerful network of research, care, and social mobilization. Along the way, Fragile X itself has been transformed as a fact.
This all may have become possible thanks to the exogenous historical shifts discussed above, but advocates had to develop a tailored set of activist repertoires and cultivate targeted alliances to turn Fragile X into a powerful clinical and social fact. Like the autism movement before them (Eyal et al. 2010), the Fragile X group united researchers, clinicians, and above all families. They learned to focus their awareness raising efforts on key constituencies like pediatricians and the psy-discipline experts who work with developmentally different children. By resisting the urge to “tell the whole world,” as one key activist put it in an interview, their targeted advocacy got more and more people tested for Fragile X. As case numbers accumulated, the Fragile X network grew and gathered new allies. Fragile X began to circulate in new fields of medical research and practice, acquiring new phenotypes and nuances along the way.
The NFXF created a database of patients, giving researchers a precious opportunity to recruit numerous subjects with a rare genetic variant in one fell swoop. The reported Fragile X phenotype began to develop or “loop” from being just one among many forms of XLMR into a far more nuanced condition strongly tied to autism and associated with a wide range of other medical, psychological, and neurodevelopmental differences. The foundation forged ties with autism organizations and embraced the idea that Fragile X could be a genetic model for autism writ large. It was only years later that they turned to the sorts of lobbying and general awareness raising that made them the envy of the field. The network now even includes mice with mutations in their FMR1 homolog gene, and with them a range of pharmaceutical experts, interests, and trials of compounds that might allay autism-like phenotypes. A stepwise process of network formation—bringing together patients, a range of new expert fields, and a host of tools, inscriptions, and allies—transformed Fragile X as a scientific fact and as a social/political cause.
This network forged a series of durable path dependencies. Ascertainment bias created a self-fulfilling association between Fragile X and intellectual disability (the Fragile X gene is still called FMR1 for “fragile x mental retardation 1,” which produces the “fragile x mental retardation protein” or FMRP), leaving many people with Fragile X undiagnosed (especially more mildly affected females who have a second FMR1 gene). It also created a powerful association with autism that would enable and constrain treatment research on Fragile X to this day.
The network created material path dependencies too. Fragile X syndrome and carrier tests were increasingly routinized and widely disseminated, and the growing availability of Fragile X patients, treatments, model organisms, and data for research studies set the stage for further changes in its status as a fact. Finally, the intensely family-oriented community that formed around Fragile X made a series of adult-onset conditions visible among carrier mothers and grandparents 6 —a finding that would transform Fragile X research and sociality in the twenty-first century. As Fragile X researchers and families literally gathered together—making anecdotal connections and taking straw polls among conference attendees—new things came into view. They began to realize that “carrier” relatives seemed to experience a range of late onset issues that have now been dubbed FXTAS (Fragile X tremor and ataxia syndrome) and FXPOI (primary ovarian insufficiency) (Mila et al. 2018). A more recent body of research points toward a range of lifelong neurodevelopmental differences among FXS carriers, increasingly known as FXAND (Fragile X-associated neuropsychiatric disorders) (Hagerman et al. 2018). This is still a developing area of research, but it has far-reaching implications when 1 in 290 to 855 males and 1 in 148 to 291 females are Fragile X carriers according to the CDC. 7
In sum, Fragile X has gone from a mere subset of XLMR to the kind of dynamic and fine-grained diagnosis that can change almost every aspect of a patient’s treatment, education, and identity. It is now a “Family Affair” (a major NFXF trope) with implications for theories of inheritance (Fu et al. 1991), the genetics of autism (Hagerman, Rivera, and Hagerman 2008), and potentially hundreds of thousands of mildly affected carrier-patients.
Over the past few decades, new conditions of possibility have seen more and more genetic variants and genomically designated syndromes emerge as highly salient facts. The cultural power of DNA, the human genome, and what have you looms large (Nelkin and Lindee 2004), even in our “postgenomic” era. Genetic testing has become far more sensitive and affordable, allowing for the discovery of countless genome variants. It is also far more routine and widely available, even as access and coverage remain highly uneven across race, place, healthcare systems, and socioeconomic status. Major databases allow clinicians quick access to detailed information about thousands of established genetic conditions; others help researchers connect far-flung cases of a novel variant and build up its clinical profile. For genetic variants that often cause life-threatening issues like heart malformations, innovations in fields like cardiology and neonatal care mean many more bearers will survive early childhood. As the “gene-for” model has failed for common illnesses, postgenomic researchers and pharmaceutical companies have increasingly turned to rare genetic disorders as the key to “fulfilling the promise of molecular medicine” (the title of an influential Annual Review of Medicine piece about Fragile X; Krueger and Bear 2011).
Patient advocacy has become an institutionalized part of the political economy of health, and a series of legal and economic reforms supporting disability rights and rare disease research have been enacted (Khosla and Valdez 2018). Of course, patients and families affected by rare variants can now go online and quickly find local communities or organizations like the NFXF. When such a community does not yet exist, patients, parents, and researchers can connect far-flung cases and jumpstart network formation using databases and social media, rather than having to somehow find each other through expert connections and serendipity. Above all, the Fragile X movement created a set of social movement repertoires that other advocacy groups explicitly draw on today. In this way, the painstaking work of forging a network around one scientific fact can become another fact’s vital condition of possibility. These sorts of exogenous factors decisively shape the terrain for knowledge accumulation and social action about any given genetic variant.
Consider the 22q11.2 microdeletion: a missing segment of a few hundred thousand DNA base pairs at site 11.2 on the long arm of the 22nd chromosome. It had been tentatively reported in the 1980s in a few patients with a very rare clinical disorder, DiGeorge syndrome, using long-standing cytogenetic techniques (de la Chapelle et al. 1981). However, by the 1990s, new technologies allowed for a reliable test that specialists could consider ordering, and the microdeletion was quickly discovered in patients with several other rare clinical disorders, as well as other people who did not exhibit telltale signs of a genetic condition (McDonald-McGinn et al. 2001). 22q11.2 deletion syndrome (DS), as it came to be known, is now associated with congenital heart defects, submucous cleft palate, ADHD, mild intellectual disabilities, schizophrenia, and a wide range of around 200 other symptoms and differences. Yet, it is also diagnosed in people who are very mildly affected—some barely at all. 22q11.2DS’s prevalence is often estimated to be around 1 in 3,000 to 6,000, although many experts think it is probably much more common (McDonald-McGinn et al. 2015), with advocates citing a figure of 1 in 1,000. 8 More recent genome-wide tests mean a 22q11.2 microdeletion can be detected even if no one ordered a test for it per se—a huge boon for rates of diagnosis when there is so much overlap and variation within and between variants, and when no one can possibly be familiar with every genetic disorder out there.
In the mid-1990s, a decade after the Fragile X group, a hybrid parent–expert “22q” support group started to take shape around another kitchen table—this time in Philadelphia. By the 2000s, they had grown into the International 22q11.2 Foundation—the central node in an international network of research and advocacy modeled on the Fragile X movement. A leading figure in 22q11.2DS research and advocacy told me they talk about emulating the Fragile X movement in almost every planning meeting, and at one of their annual conferences there was a whole session dedicated to “Lessons from Fragile X.” Sure enough, just like Fragile X before them, 22q11.2DS researchers and advocates have built a highly active foundation, created strong ties to researchers and pharmaceutical companies interested in overlapping common conditions like schizophrenia and anxiety (Gur et al. 2017), developed a series of management guidelines published in major journals and promoted by 22q foundations, and spearheaded an international network of specialist clinics. 9 22q11.2DS went from a topic confined mostly to cardiology and psychiatry to take on new life as a fact in other fields—endocrinology, pediatrics, and many other clinical specialties, as well as even further afield in special education, child psychology, and so on. The 22q11.2DS diagnosis provides entrée into a world of foundations, Facebook groups, conferences, specialist clinics and referrals, 22q summer camps, and much more. In this way, the 22q11.2 microdeletion has looped from a putative “gene-for” a very rare clinical disorder to a borderline common condition, 22q11.2DS, complete with a sprawling social world and a highly complex phenotype that sometimes blurs the boundary between normal and pathological. It has become the sort of scientific, clinical, and social fact that can transform a patient’s life.
Advocacy organizations like the NFXF and the International 22q11.2 Foundation can raise funds and lobby public agencies for research and care resources, and they leverage their moral authority and gatekeeper status to direct research toward the kinds of issues that patients and their families care most about (Panofsky 2011; Terry et al. 2007). They also create new forms of visibility (Wailoo 2001) that change a variant-as-fact. In some instances, community members themselves recognize common features in people with the genetic variant, like the aversion to eye contact in Fragile X or the striking split between verbal and performance IQ in 22q11.2DS, which then become part of the variant’s formal phenotype when they are published in biomedical journals.
Experts and advocates often work together to develop new resources that become a key part of these networks. Patient registries and repositories like the NFXF Data Repository 10 help facilitate, guide, and scale rare disease research, and initiatives like the Fragile X Clinical and Research Consortium (FXCRC) foster a network of specialist clinics to provide tailored care while also creating the easily-accessed population of research subjects that make large studies of a rare disease possible. 11 These networks also develop “immutable mobiles” (see earlier discussion of Latour 1988) to extend the power of the network. Published clinical management guidelines, factsheets, and other resources offered by groups like the NFXF or the International 22q11.2 Foundation 12 are crucial tools: parents can use them, for example, to secure referrals and resources from local healthcare providers and schools who are unfamiliar with rare genetic disorders, or to help explain the meaning of a genetic diagnosis to their children. These objects and inscriptions are an integral part of the networks assembled around genomic variants, and they have a profound impact on the meaning of a genetic diagnosis across an array of key sites.
Finally, let us return to XYY. Thanks to groups like the Association for X and Y Chromosome Variations, or AXYS, Trisomy X and XYY are very different facts than they were when they represented the cutting edge of human genetics in the 1960s. XYY has made a particularly striking comeback. After decades in the scientific wilderness, it is once again an important diagnosis—one that bears important points of continuity with the XYY of the 1960s even as it has undergone a dramatic transformation. While acne and increased stature have remained constants, aggression, deviance, and “mental subnormality” have been replaced by an increased risk of ADHD or high-functioning autism and an average of around 10 to 15 IQ points lower than unaffected siblings. 13 New programs of XYY research, treatment, and support in areas like special education are underway. XYY is more likely than ever to be detected, both through expanded genetic testing for neurodevelopmental differences and the new wave of noninvasive prenatal screens that bring with them the option of selective abortion (in places where it remains legal).
Even as stigmatizing tropes about the “criminal chromosome” linger, a dramatically different profile has emerged. AXYS have produced materials like “For EXtraordinarY BoYs: A Guide to 47,XYY” 14 outlining XYY’s spectrum of mild neurodevelopmental challenges and differences. They promote a children’s storybook called Jack and His Extra Y (Colvin 2014) 15 that explains what genes and chromosomes are and how XYY, while sometimes having no effect at all, makes Jack just a little different: “He is not ashamed of his extra Y. That is just the way he is. Jack was made with blond hair, green eyes, smelly feet, and an extra Y. . . . He looks just like the other boys. But inside his brain, sometimes his extra Y chromosome makes him feel different. Sometimes having an extra Y can make things harder for him, like reading or doing schoolwork.” Under new conditions of possibility, and with a new network assembled around it, XYY has been transformed as a scientific and social fact.
The power of these networks is perhaps best captured via a comparative perspective. To this day, variants like XYY, Fragile X, and the 22q11.2 microdeletion—and many more besides—mean very different things in different fields, institutions, places, and especially across various dimensions of inequality like social class, place, and race/ethnicity. Indeed, the long-standing underrepresentation of people of non-European ancestry in genomic databases often makes it hard to determine whether some single-gene variants are even pathogenic in non-white patients (Fatumo et al. 2022). For these and many other reasons, both rates of diagnosis and the ability to use a genetic diagnosis to inform treatment and care are all highly concentrated among privileged populations. Most strikingly of all, the vast majority of variants remain very limited as scientific or social facts, not necessarily because of anything related to their rarity, pathophysiology, and so on, but because they have not been the subject of sustained network formation. But when a diverse network of expertise and social mobilization is organized around a genetic variant, even a very rare one, it begins to loop and develop as a fact.
Yet, genetic variants and conditions are still indelibly shaped, as facts, by contingent events and developments from the past. Perhaps the most profound path dependency comes from the various forms of ascertainment bias that limit and guide what we know about them. Clinicians often only think to test for a variant in people with the cardinal signs—for example, a specific heart defect in the case of 22q11.2DS or serious developmental delays for Fragile X. That means we routinely miss people with different clinical features and ranges of abilities. In this way, the phenotypes reported in early research on a variant create durable path dependencies that cannot be overcome without a great deal of time and investment. Over time, incidental ascertainment and familial testing teaches us that many of these variants are also found in people who are less severely affected, affected in other ways, or perhaps barely affected at all, but the differentials for genetic testing are still geared toward long-standing clinical profiles. There is no easy way out of this riddle. As Timmermans and Buchbinder (2013) have shown, presymptomatic screening can begin to resolve ascertainment bias, but often at the cost of creating “patients in waiting” and unleashing a series of far-reaching dilemmas about the true meaning and status of a genetic diagnosis—not to mention the costs of follow-up testing, analysis, surveillance, and care.
In other instances, a newly discovered variant may stand in a complex relationship to the existing clinical nosology, creating confusion and further ascertainment bias. In the case of the 22q11.2 microdeletion, the rare clinical syndromes in which it was initially discovered have lingered for decades, leading patients who test positive to variously receive 22q11.2DS, DiGeorge syndrome, velocardiofacial syndrome (VCFS), and other diagnoses even though the field considers them synonymous. This situation led to an acrimonious, years-long dispute between a group of leading researchers and figures in the field who were dedicated to the older VCFS name with their VCFS Education Foundation and the group that formed the International 22q11.2 Foundation. This was all extraordinarily frustrating for patients and parents, and a major drain on a rare disease network that already faced steep hurdles. It ultimately led 22q advocates to embark on a years-long “Same Name Campaign.” 16 In sum, epistemic path dependencies can create thorny challenges for rare disease researchers and advocates as they work to assemble new knowledge and a larger patient population.
These reinforcing biases are easily overlooked, precisely because they so thoroughly shape the variant in question as a scientific fact. Indeed, they also create material path dependencies: our delineation of “the” variant itself across successive generations of genetic testing technologies, the population who has been diagnosed, the community of people whose identity is tied to it, the institutional settings where it circulates as an object of research and practice, the gene panels it is included on or excluded from, the data and biospecimens found in patient registries and biobanks, and many other elements. The biomedical literatures, clinical guidelines, and patient-facing materials produced by these networks create self-fulfilling prophecies, shaping what clinicians look for and what parents expect from their children. When model organisms are based on a particular understanding of a mutation—its most salient phenotype, proteomic mechanism, the “key gene” in a chromosomal anomaly, or its status as an “autism gene”—that will set the scope for pathophysiological research and perhaps even pharmaceutical development moving forward. Finally, the highly contingent decision to include a genetic variant on a prenatal testing panel has huge implications for the way it is understood, even raising the question of whether it is “avoidable” or compatible with a “life worth living” (Lippman 1991; Parens and Asch 1999). The recent inclusion of variants like XYY and 22q11.2 in the new generation of commercial noninvasive prenatal genetic screening kits may have far-reaching demographic implications: who is identified with the variant and who is born with it, with everything that entails for the associated networks of expertise, social mobilization, and power. These and other material contingencies create durable paths for a genetic variant’s development status as a fact.
Most importantly of all, the establishment of a patient advocacy movement itself involves a healthy degree of contingency—when, where, and especially if it happens. A whole universe of genetic variants and genomically designated conditions are well established as facts in the literature, but have not gone much beyond that. These variants have not been taken up by support groups and advocates, and as a result they have not been intensively studied or developed beyond a thin clinical profile. Even when a variant is clearly pathogenic, consistently serious, and not especially rare, building this sort of hybrid network is a daunting task. That is why so many genetic variants remain poorly understood and rarely diagnosed. Yet, even extremely rare point mutation conditions like the ADCY5 or NGLY1 deficiencies can be catapulted into public awareness and major scientific investment (Anon n.d.; Mnookin 2014; Yong 2015). With today’s conditions of possibility, just a few parents with the right combination of social, cultural, and economic capital can assemble the sort of network that turns a rare genetic variant into a dynamic biomedical and social fact. In these and many other ways, the contingent actions of a few people can lead to programs of research and mobilization that irrevocably change genetic variants as facts.
Discussion
Our lives have all been upended over the past few years by a dynamic new scientific fact: the SARS-CoV-2 virus and the COVID-19 disease it causes. Its disruptive, deadly effects have been felt all over the world. This is no matter for your garden variety, anti-realist constructivism. On the contrary, it is precisely a careful interrogation of this new reality that compels us to look far beyond SARS-CoV-2 itself. This novel coronavirus has very different contours as a scientific fact in different places—differences that depend on all manner of preexisting conditions of possibility. Our very understanding of it as a zoonotic virus—never mind one whose history, function, vulnerabilities, and variants can be revealed by looking at its ~30,000 base pair genome—depends on a series of revolutionary developments in biology and medicine. Prior factors like prevailing social norms; urban density; governance structures and public health infrastructures; demography and the prevalence of various preexisting conditions; and divergent experiences with SARS CoV-1 (Eyal 2021) shape fundamental aspects about the virus-qua-fact, not least its prevalence (real and recorded), attack, and mortality rates. The networks of expertise, social mobilization, and power deployed in response have been notoriously varied, even among advanced-economy nations with large scientific capacities. Here in the United States, we saw these conditions of possibility and the complex, often troubling networks of dis/mis/information and practice that coalesced around COVID-19 play out all too clearly (Prasad 2022). Finally, our divergent responses to SARS-CoV-2 will continue to shape the future for this virus, others that follow, and perhaps many other issues in medicine, public health, and beyond. It has never been clearer that sociologists need to grapple with the way scientific facts unfold over time.
Reiterated fact-making can help us grasp how many different sorts of scientific facts are made and remade according to prevailing conditions of possibility, the different networks of expertise, social mobilization, and power assembled around them, and the path dependencies that enable and constrain the way facts develop and get used. It is a flexible tool. For example, this approach has recently been used to analyze the history of drought in California, which started off as a social fact before being painstakingly rendered into a hybrid social/scientific fact from the mid-twentieth century on (see McInnis 2022; McInnis and Navon 2024)—precisely the opposite journey between scientific and social facticity that played out in this article’s case studies. Reiterated fact-making helps us understand the shifting conditions, networks, and path dependencies that shape every aspect of drought in California: what it is, when and where it happens, and its various effects. The framework can be readily applied to established facts, contested facts, or cases of “undone science” (Frickel et al. 2010; Hess 2016)—indeed, this article’s case studies have taken on all three statuses at various points in their careers as facts. Whether it is COVID-19, pathogenic variants, or drought in California, reiterated fact-making allows for comparative-historical explanation not only of what we do with scientific objects, but also how we define and understand them—what they are as facts.
A couple points of clarification are in order. First, scientific facts are often “enacted” (Mol 2002) in divergent ways across different disciplines and sites. Even a single fact may “contain multitudes” (Whitman [1855] 2005:43), combining insurmountable internal tensions between coherence and polysemy, continuity and transformation. A fact is never one fixed thing, but a shifting amalgam that simultaneously changes and becomes more real whenever it is represented, used, or acted upon in new fields of research or practice. XYY, Fragile X, and 22q11.2DS mean very different things in different sites and fields. In places untouched by their respective networks, they mean little or nothing at all. Nevertheless, despite the uncertainties that are usually so present at the beginning of their scientific careers—not to mention endless debates about details and responses—facts are often rendered stable enough that no one questions whether they are important things in the world. If a fact has a coherent, continuous lineage as a thing—even if that continuity represents a major sociotechnical accomplishment requiring extensive stabilization and “calibration” (Hacking 1998:98–101)—then it may be well worth tracing across time and space. It is precisely this combination of mutability and stability that can make a scientific fact very powerful. Reiterated fact-making therefore must be rigid enough to stay steadfastly focused on a fact as the object of comparative-historical casing while also allowing for far-reaching variation and change.
Second, it almost goes without saying that any given study will never unpack everything there is to say about a scientific fact—it all depends on what one is trying to learn or explain. Indeed, our case studies were very much focused on the United States and other Western countries. We will clearly need to identify different sets of factors, actors, objects, and processes to explain the social histories of these genetic variants and conditions as they become more salient facts in other places over the coming years.
That said, we can use reiterated fact-making not only to explain how and why facts change over time, but also as a targeted analytic vantage point for getting at broader topics of sociological inquiry. After all, scientific facts shape just about every domain of social life. By carefully unpacking the conditions of possibility, networks, and path dependencies that shape scientific facts, we can generate orthogonal support for existing findings and generate new ones. For example, using reiterative fact-making to trace genetic variants confirms that diagnostic expansion drove the so-called autism “epidemic” (Eyal et al. 2010; King and Bearman 2009), but it also shows us how that same process changed the genetic makeup of the autism population and therefore the way we understand autism’s pathophysiology (Navon and Eyal 2016). It confirms Epstein’s (1996) seminal finding that hybrid lay experts/activists can remake the terrain for medical research, but it also shows how they can help drive a “revolution” (Astle et al. 2022) in the way some fields classify illness and difference in the first place. Finally, it helps us understand how it is not just that rates of genetic diagnosis are lower in underprivileged and BIPOC populations, but that having a genetic variant can mean very different things depending on the resources a patient and their family can access. In short, reiterated fact-making can shed new light on broader power dynamics, inequalities, and processes of scientific innovation and social change.
Finally, it is important to emphasize once again that this article shamelessly draws on a rich legacy of scholarship in the historical and social studies of science at almost every turn. Nevertheless, reiterated fact-making provides an analytic framework for sociologists seeking to make comparative-historical sense of scientific facts. In this way, by rigorously centering facts in a narrative explanation, we can bring new insight to topics that have mostly fallen to STS scholars. We can truly explain how scientific facts emerge, combine continuity and transformation as they interact with the world around them, and shape people’s lives.
Footnotes
Acknowledgements
I am very grateful to Alberto Cambrosio, Dan Driscoll, Gil Eyal, Jeffrey Haydu, Ilana Löwy, Haley McInnis, and Stefan Timmermans—as well as the anonymous reviewers and ASR editors—for their invaluable feedback on this paper. I would also like to thank colleagues who attended talks and asked incisive questions at ASA, SSHA, and UCLA Sociology’s Theory and Research in Comparative Social Analysis seminar.
Funding
This study was funded by the UCSD Hellman Fellowship, the UCSD Academic Senate General Research Grant program, the Robert Wood Johnson Foundation Scholars in Health Policy Research Program, and the Binational Science Foundation (grant number 2010175).