
Abstract
A highly efficient Monte Carlo (MC) algorithm is developed for the numerical simulation of aerosol dynamics, that is, nucleation, surface growth, and coagulation. Nucleation and surface growth are handled with deterministic means, while coagulation is simulated with a stochastic method (Marcus-Lushnikov stochastic process). Operator splitting techniques are used to synthesize the deterministic and stochastic parts in the algorithm. The algorithm is parallelized using the Message Passing Interface (MPI). The parallel computing efficiency is investigated through numerical examples. Near 60% parallel efficiency is achieved for the maximum testing case with 3.7 million MC particles running on 93 parallel computing nodes. The algorithm is verified through simulating various testing cases and comparing the simulation results with available analytical and/or other numerical solutions. Generally, it is found that only small number (hundreds or thousands) of MC particles is necessary to accurately predict the aerosol particle number density, volume fraction, and so forth, that is, low order moments of the Particle Size Distribution (PSD) function. Accurately predicting the high order moments of the PSD needs to dramatically increase the number of MC particles.
1. Introduction
Population balance equations (PBEs), describing particulate entities conservation, have applications in many branches, such as aerosol dynamics [1, 2], polymerization [3], and so forth. Here, the discussion mainly focuses on aerosol dynamics. Aerosol refers to a colloid suspension of fine solid particles or liquid droplets, which can be found in clouds, air pollution such as smog and smoke, soot in combustion flames, cement dust, and so forth. Analytical solutions to the PBEs are available for only a few specific cases [4, 5]. The most employed general method and its variants for solving the PBEs is based on dividing the particle size domain into sections as developed by Gelbard et al. [6]. Usually, these direct discretization methods work well for either coagulation or condensation dominated aerosol dynamic processes, but they encounter various problems [7] when dealing with combined nucleation, coagulation and condensation, for example, numerical error in specifying a given distribution in a section, non-positiveness and non-conservation of mass and number concentration due to numerical diffusion.
Other than solving the discretized PBEs, a widely used alternative approach has been the method of moments, which solves a group of moments equations derived from the PBEs. These moments (generally several low order moments) provide important information on the PSD function, that is, number density, volume fraction, polydispersity, and so forth. However, owing to the nonlinearity of the PBEs, the governing equations for lower order moments contain higher order moments, which are not closed after cutting off. Various closure methods have been developed, such as unimodal log-normal approximation [8], quadrature/direct quadrature [9, 10], interpolative closure [11], Taylor expansion [12–14], and so forth. Although the method of moments is computationally highly efficient, the complexity of the closure makes it difficult to accommodate complex physical models for aerosol dynamics with high flexibility.
Monte Carlo (MC) simulation does not “solve” the PBEs, but to mimic the evolution of aerosol particles through a stochastic particle system. Modeling coagulation by stochastic particle systems was introduced by several authors [15–17], which is usually called Direct Simulation Algorithm (DSA). Eibeck and Wanger [18] proposed a stochastic algorithm that models the weighted Smoluchowki's coagulation equation, which exhibits lower variance than the DSA for predicting the tail of the PSD. There are modifications to the DSA aiming for higher computational efficiency. The majorant kernel method [19, 20] adopts an acceptance-rejection technique to speed up the simulation. The increased efficiency highly relies on finding a “good” majorant kernel. The so-called τ leaping method [21, 22] is an approximate accelerated stochastic simulation, which enables multiple coagulation events at a time. The extension of the DSA to model general aerosol dynamics (nucleation, surface growth, etc. also included) goes to diverse directions, time or event driven, constant volume or number MC simulations [23–25]. Most of these algorithms share the same idea [26] to randomly choose a aerosol dynamic process (either nucleation, or coagulation, or surface growth) at a time to determine the aerosol particles evolution. Other than selecting all processes randomly, Patterson et al. [27] proposed the linear process deferment algorithm, which separates the surface reaction from other dynamic processes, and models the surface reaction independently for some steps, and then includes the other processes. Their idea to accelerate the simulation is similar to the τ leaping method in some sense.
In this paper we propose a parallel hybrid algorithm of stochastic simulation and deterministic integration for solving the PBE, which is called Operator Splitting Monte Carlo (OSMC). OSMC adopts operator splitting techniques (similar idea is also found in Patterson et al. [27]) to separate coagulation from other processes. Coagulation is simulated by the DSA, but all other processes are modeled by deterministic integration of their dynamic equations. Stochastic simulation is much more efficient than directly solving the Smoluchowski integro-differential equation with traditional discretization [23]. However, using stochastic method for nucleation and surface growth is far more expensive than their corresponding deterministic solution methods. The combination of stochastic and deterministic methods makes OSMC very efficient and flexible for aerosol dynamics simulation. OSMC is used to simulate a series of well-chosen testing cases. Detailed comparison with analytical and/or other numerical solutions is provided. The paper first introduces the OSMC algorithm, then discusses the simulation results of the testing cases.
2. Methodology
2.1. Governing Equations
The evolution of the PSD function, n (v,t), is described by the PBE (also called the general dynamic equation) [1]

where

that is, integrating the time that a fluid parcel travel to the spatial position
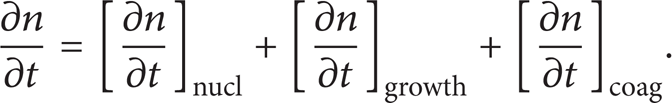
This work focuses on solving (3) with the OSMC algorithm. The convection of aerosol in the flow field is not considered here. There are many practically interested conditions, where the flow field can be considered as steady and one dimensional, for example, burner stabilized flames and counter flow diffusion flames. Then it is very easy to integrate (2) to build up a one-to-one mapping between the time and spacial coordinate. Hence the convection can be readily assimilated in the model equation (3). For general cases of aerosol evolution in three dimensional flows, it is possible to use the Lagrangian particle scheme to simulate the aerosol evolution along Lagrangian trajectories [28, 29]. Then (3) still applies along the Lagrangian trajectories. So the model equation (3) and the subsequent OSMC algorithm have very broad application.
The aerosol dynamic processes on the right hand side describe the interactions among molecules (or cluster of molecules) and particles. Nucleation is the process that dozens or hundreds of molecules form a stable critical nucleus (particle (Droplets and particles are treated as synonyms in this article)). It is usually modeled by the classic Becker-Döring theory [30] or its variant self-consistent correction theory [31]. Surface growth includes physical condensation and surface chemical reactions, which involves interactions between gas phase molecules and an aerosol particle. Condensation rate in the free molecule regime (i.e., when aerosol particles are smaller than the mean free path of the gas) is proportional to the surface area of particles, and it is proportional to the diameter in the continuum regime (i.e., when aerosol particles are bigger than the mean free path of the gas) [1]. An approximate interpolation formula for the entire range has been proposed by Fuchs and Sutugin [32]. Surface chemical reactions are too broad to address. A typical example in soot formation in combustion flames is the hydrogen-abstraction/carbon-addition soot growth mechanism [33]. Coagulation is the process that two particles coalesce to form a bigger particle. Here, coagulation may also refer to aggregation. The coagulation dynamics is described by the well-known Smoluchowski's equation

The collision kernel function β(v,u) describes the rate at which particles of size v coagulate with particles of size u. Analytical solutions for the Smoluchowski's equation are known [34] when β has one of the following three forms, (i) β(v,u) = 1, (ii) β(v,u) = v + u, and (iii) β(v,u) = vu. Practically, more complex physical models for β are developed. When particle diameter is smaller than the mean free path of air (the free molecule regime), β can be expressed as [1]
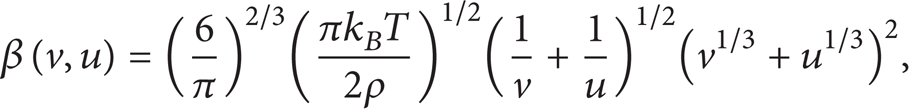
where v and u are the volume of the (spherical) colliders, T is the temperature, k B is the Boltzmann's constant, and ρ is the density of particles.
2.2. Operator Splitting
The governing Equation (3) contains very different physical processes. Operator splitting is an efficient and powerful method to solve such evolution equations [35]. In operator splitting method, the time interval [0,t] is descritized as usual, and let δt i denote one time step. The solution n(t) can be constructed by classic methods, such as the forward Euler method. Other than to integrate all dynamic processes together in one step (denoted as exp(δtX)), operator splitting method separates the integration to multiple steps, such as


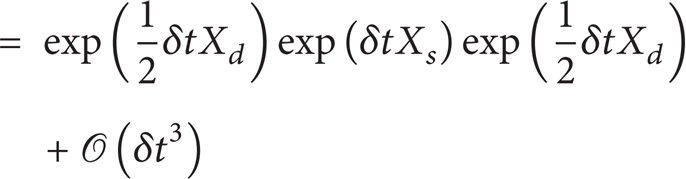

where X d denotes nucleation and surface growth processes, which is solved by deterministic integration method, and X s denotes coagulation, which is modeled by the Marcus-Lushnikov stochastic process [36]. Equations (6a) and (6b) are first order Lie schemes (the global error at t is 𝒪(δt), while is one order lower than the local error in a step), (6c) and (6d) second order Strang schemes. Figure 1 gives a schematic view of the operator splitting techniques.

Operator splitting schemes. (a) first order Lie scheme; (b) second order Strang scheme. Inverting the order of X d and X s gives the other Lie and Strang schemes, respectively. X s denotes MC simulation for coagulation, X d denotes deterministic integration of the other aerosol dynamics.
2.3. MC Simulation of Coagulation
The stochastic algorithm of Gillespie [36] is chosen to simulate coagulation. In essence, the simulation algorithm is as follows:
Step 1. Specify initial values (volume, surface, diameter etc.) for N particles, determine the simulator size V = N/n0 (where n0 is the initial number density), and evaluate the pair coagulation rate C ij = β(v i ,v j )/V, (i = 1,…, N–1, j = i + 1,…, N).
Step 2. Generate a random time τ at which two particles coagulate, where τ satisfies the Poisson distribution

Here C0 is the summation of all pair coagulation rate,
Step 3. Randomly choose two particles to coagulate according to the distribution P(i,j) = C ij /C0.
Step 4. Update N (one particle less) and C ij , and repeat Steps 2 and 3, until the accumulated random time τ is larger than the simulation time tstop.
Gillespie [36] provided three methods to realize Step 3. The partial conditioning method therein is adopted here, which is usually the most efficient and easy to implement for parallel computing. The calculation of C0 has complexity 𝒪(N2) and is the most computationally expensive step in the algorithm. However, there is simple way to update C0 by substracting from its current value the contributions of the two particles that collided and adding the contributions from the newly formed particle. Those operations have complexity 𝒪(N) and have negligible costs compared to the computation of C0. It is worth pointing out that the evolution of C0 can be computed by the majorant kernel method [19, 20], which is numerically more efficient than computing C0 directly. However, in our algorithm C0 is computed directly only at the every beginning, then C0 is updated by the simple and efficient scheme. Hence the majorant kernel method may not outperform our simple updating scheme.
2.4. Deterministic Integration of Nucleation and Surface Growth
Nucleation generates particles with a specific size, which does not depend on the existing aerosol particles. Although there are various nucleation theories for various kinds of aerosol, such as droplets [30, 31], soot particles [37], nucleation rate is generally a function of temperature and nucleated vapor concentration (or polycyclic aromatic hydrocarbon for soot). For a given nucleation model, the nucleation term in (3) can be easily integrated to give the number density of nucleated particles. Surface growth may refer to physical condensation process or surface chemical reactions. Other than on the temperature and surrounding vapor concentration, surface growth rate usually also depends on the size of aerosol particle. In the OSMC algorithm, the size of aerosol particle (modeled by MC particles) by is known. Surface growth term can also be readily integrated.
A self-adaptive fifth order Runge-Kutta method ([38], rkqs subroutine) is used to integrate the dynamic equations for nucleation and surface growth within a discrete time step δt. This choice adds flexibility to control the integration error to a given lever. Numerical experience shows that one-step explicit Euler integration is usually sufficient, that is, X d (t + δt) = X d (t) + δt(dX d /dt).
2.5. Full Algorithm Including All Aerosol Dynamics
Figure 2 shows the flowchart of OSMC including all aerosol dynamics. Only the first order operator splitting (6a) is sketched in the figure.
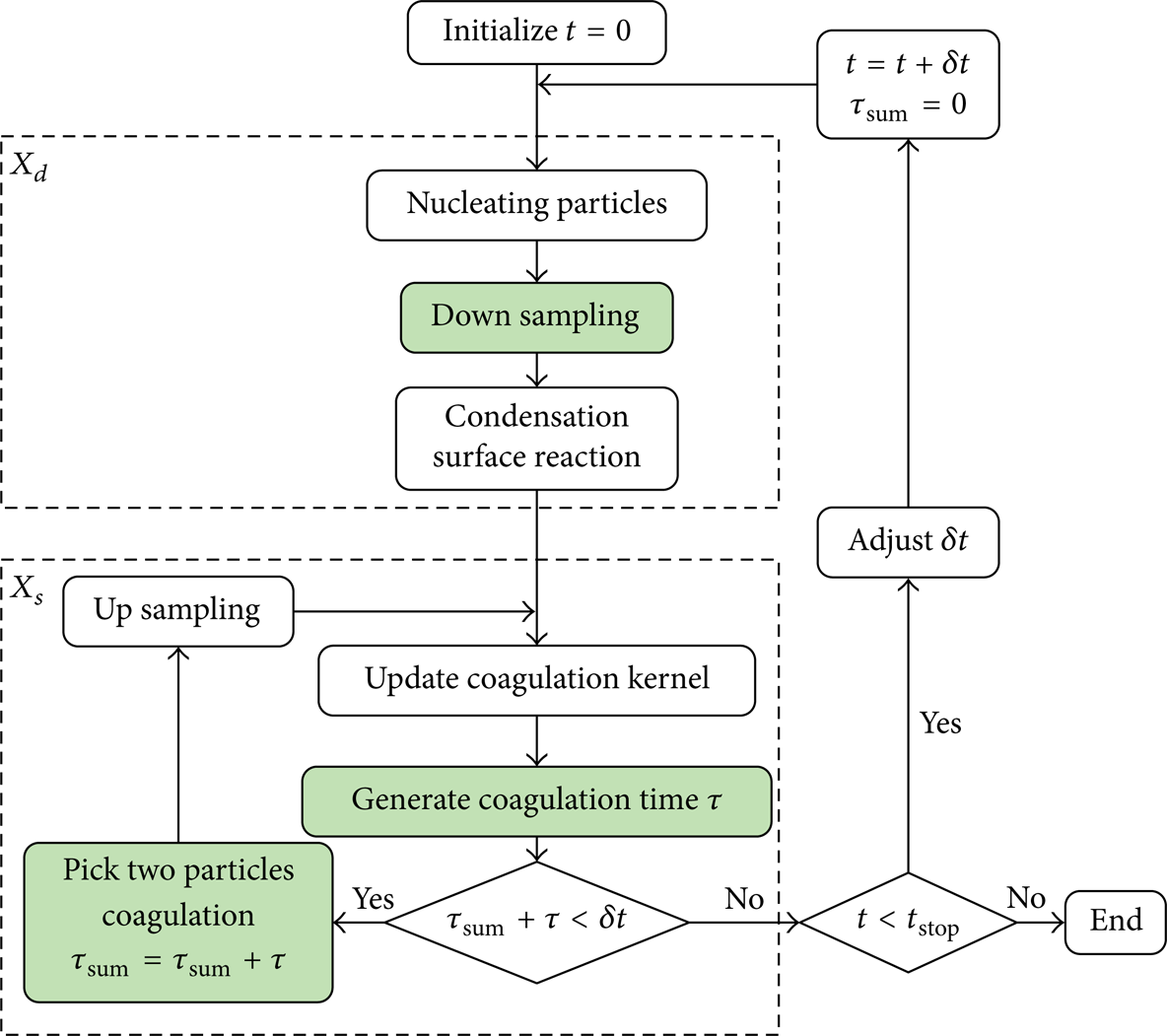
Flowchart of the OSMC. Filled blocks denote stochastic steps. Here up-sampling is limited to the doubling method, hence no stochasticity is introduced.
At time t = 0, the simulator is initialized with given parameters, that is, number of MC particles N, simulator size V, and initial PSD. The simulator size is determined as V = N/n0, where n0 is the initial number density. If simulation starts from an empty case, that is, n0 = 0, then the simulator is initialized with N particles of the same size for simplicity (the real value of the size is immaterial to the simulation results), and the simulator size is set to a huge number, say V = 1010 m3, which renders a tiny particle number density to approximate the condition n0 = 0. If simulation starts from a case with a specific PSD, then the size of the N simulation particles is randomly assigned to satisfy the initial PSD. There are various convenient ways to generate random number to satisfy a given distribution [39], although they are usually not necessary in Monte Carlo simulation of aerosol dynamics, since mostly the simulation starts from an empty case.
In the particles nucleation step, the simulator size V is adjusted to reflect the change of particle number density due to nucleation. Nascent nucleated particles are added to the pool of MC particles. If the total number of MC particles exceeds the maximum allowable value, then down-sampling is performed, that is, exceeding MC particles are randomly removed from the pool to satisfy the number constraint. And then every MC particle undergoes the surface growth process according to its growth rate (usually depends on its size).
The coagulation simulation process (those steps grouped in the dashed bounding box X s in the flowchart Figure 2) shows how to implement the stochastic algorithm of Gillespie [36]. Updating coagulation kernel is to calculate the pair coagulation rate C ij = β(v i ,v j )/V, (i = 1,…, N–1, j = i + 1,…, N). The random coagulation time τ is generated according to (7). The comparison statement τsum + τ<δt is to judge whether the time for two particles to coagulate is still admissible within the discrete time step δt. Here τsum is the accumulated coagulation time in the coagulation step, which is initialized to zero before a coagulation step begins. If the comparison statement is true, two particles are selected (Step 3 in the above algorithm) to perform coagulation, and the number of MC particles decreases by one. The subsequent up-sampling step is to keep the number of MC particles above a given threshold to avoid severe stochastic error.
The deterministic integration step X d and the stochastic simulation step X s can be organized according to the operator splitting techniques discussed in Section 2.2. The effects of different operator splitting will be reported elsewhere. One general conclusion is that the second order Strang scheme (6c) is optimal in considering the computational efficiency and precision.
A few important points should be addressed in the implementation of the algorithm. Within a time step δt, nucleation dumps a number of new particles to the particulate simulation system in order to reflect the change of number density in physical space. The change of number density can be any real value, but the number of newly dumped particles, Ndump, must be integer. There is inevitably a round-off error for the number density nearly 1/Ndump. When Ndump is big, the round-off error is small. However, Ndump is directly proportional to the time step δt and the nucleation rate, which means that Ndump can be very small or even zero under the conditions of every small δt or nucleation rate. To avoid the severe round-off error under these conditions, a correction to Ndump is developed. First, set
Another critical issue is how to control the number of MC particles during the simulation, since nucleation and coagulation may cause the number up or down to unappropriate values. There are a few popular methods in literature [40, 41], all of which are actually different ways of re-sampling the PSD during the simulation. Re-sampling contains up-sampling and down-sampling, either adding or removing particles. After re-sampling, the volume of the system V should also be rescaled accordingly to keep the particle number density unchanged before and after the re-sampling.
Smith and Matsoukas [41] used a constant number scheme for the simulation of coagulation, which dumps a new particle to the MC particle pool after every coagulation (when the particle number is decreased by one). Another popular scheme is the “topping up” method [40], which increases the number by a factor when it reaches the lower limit. A special case of the “topping up” is the doubling method, that is, when the particle number decreases by half, then duplicates all the particles. In this doubling method, no stochastic error is introduced. Maisels et al. [26] used a doubling-halving scheme to simulate nucleation and coagulation, when the particle number decreases to half of N, then double all the particles; when particle number increases to 2N, then remove every other particle. Maisels et al. [26] also extended the constant number scheme [41] to model nucleation and coagulation, and compared the results with those from the doubling-halving scheme. They found that both schemes give comparable results (with regard to accuracy, random fluctuation etc.), except that the fluctuation of particle volume fraction given by the constant number scheme is several orders higher than that by the doubling-halving scheme.
In our simulation, the number of MC particles is set within the range from N/2 to N. When the number decreases to N/2, all the particles are doubled. When the number exceeds N, it is down-sampled to N. This treatment has a few advantages over the constant number and the doubling-halving schemes. Obviously, it is computationally more economic that the doubling-halving scheme since it avoids particles increasing to 2N when computational cost quadruples. On another aspect, it has much less variance than the constant number scheme. In simulations including coagulation and nucleation, it is found that the constant number scheme shows very large variance when coagulation and nucleation rates are comparable. Because under this condition, large portion of the PSD would be randomly removed/duplicated incessantly. However, in the present doubling scheme, no re-sampling is needed when the particle number stays between N/2 and N. Therefore, much less uncertainty is introduced because of less frequent re-sampling.
During re-sampling it requires to randomly select m particles from existing N particles. There are two ways to select the m particles, either repeatable or non-repeatable. Repeatable selection is rather easy to implement as it only needs to generate m random integers all lying between 1 and N. However, in a testing MC simulation it is found that the variance of repeatable re-sampling is four times bigger than the non-repeatable case, under the worst condition m = N/2 (If m>N/2, the inverse selection can be adopted.). Non-repeatable selection requires all m particles are distinct. It is not efficient to repeatedly draw a particle randomly and check whether it has been selected before until m particles are chosen out. Durstenfeld [42] proposed a very smart and efficient algorithm for non-repeatable selection. We re-introduce the algorithm here, since it is very easy to follow. Given an integer sequence from 1 to N, first, randomly pick one number to permutate with the last one. Next, repeat the permutation in the fist step for the beginning N–1 numbers. And next for N–2, and so on for m steps. The last m numbers in the sequence are now randomly selected from the original sequence.
2.6. Parallelization
Computational storage and speed limit the applicability of MC to simulations with relatively small number of particles [43]. Parallel computing can greatly alleviate the storage and speed limits. For the simulation of aerosol dynamics, only coagulation requires to evaluate the interaction between two particles. Hence, it is only necessary to develop parallel scheme for coagulation. Other aerosol dynamics are treated with the same manner as in serial simulation, except that the information about the total particle number density and the simulator volume should be gathered and broadcast to every processor.
Section 2.3 has introduced the partial conditioning method of Gillespie [36], who also introduced other two methods, full conditioning and first-coalescence. But the partial conditioning method is usually the most computationally efficient, and the most convenient for parallel computing.
The Message Passing Interface (MPI) is used in the parallel computing. Every computing processor has N p particles assigned initially, and there are totally NCPU processors. The total particle number is N = N p · NCPU. Every processor needs to communicate with other NCPU-1 processors. The total communication channels among the processors are (NCPU-1)NCPU/2 (since communication is reciprocal). However, in the current parallel algorithm a processor deals with only N b = (NCPU-1)/2 channels, not to communicate with all other NCPU-1 processors. The communication between any two processors are accomplished through either the direct channel (if has) or indirect multi-channel. A bookkeeping table is created to record the communication structure, that is, sending to and receiving from which processor. Here, NCPU is required to be an odd number, otherwise the computational load cannot be evenly distributed among the processors. This peculiarity would usually suffer a little waste of computational resource, since computer hardware is always constructed with even number processors.
Every processor has a buffer array of size N b . The element buffer of the array holds all the information received from a specific processor, which is determined in the bookkeeping table. In evaluating the matrix C ij , every processor is assigned an equal size block from the matrix. The information for i and j particles is either both available locally, or one is available locally and the other could be find in the buffer sooner or later. The case of two alien particles has been ruled out during the construction of the bookkeeping table. This process is guaranteed to work, since all processors keep sending and receiving information (non-blocking communication) until all processors finish the evaluation of C ij . After that, the global information (summation of all C ij elements and the maximum element of C ij ) is gathered and broadcast to every processor.
The key point in this parallel algorithm is the use of the bookkeeping table, which evenly distributes computational load and launches communications simultaneously among processors. Its efficiency is tested in the next section.
3. Results and Discussion
3.1. Parallel Computing Efficiency
Generally, coagulation is the most computationally intensive part in all aerosol dynamics. Besides, only coagulation involving two particles interaction and requires message communication between computing processors in parallel computing. Therefore a testing case of coagulation in the free molecule regime is simulated by the OSMC to investigate the parallel computing efficiency under an eight-core workstation and a computer cluster. The testing case was originally used by Frenklach and Harris [44] to validate their moments method. Initially all particles are mono-dispersed with diameter 1.5 nm, and density 1800 kg·m−3. The particle number density is 1018 m−3. The temperature is kept constant 1800 K. The simulation time is tstop = 4.5 ms.
The coagulation scheme (Section 2.3) requires N2/2 operations to initialize the symmetric matrix C ij . Every coagulation event only needs 𝒪(N) operations. On the other hand, the average time during two subsequent coagulation is proportional to 1/N, which means that increasing N will cause coagulation events more often within the time interval [0,tstop]. Hence the total operations should be proportional to N2.
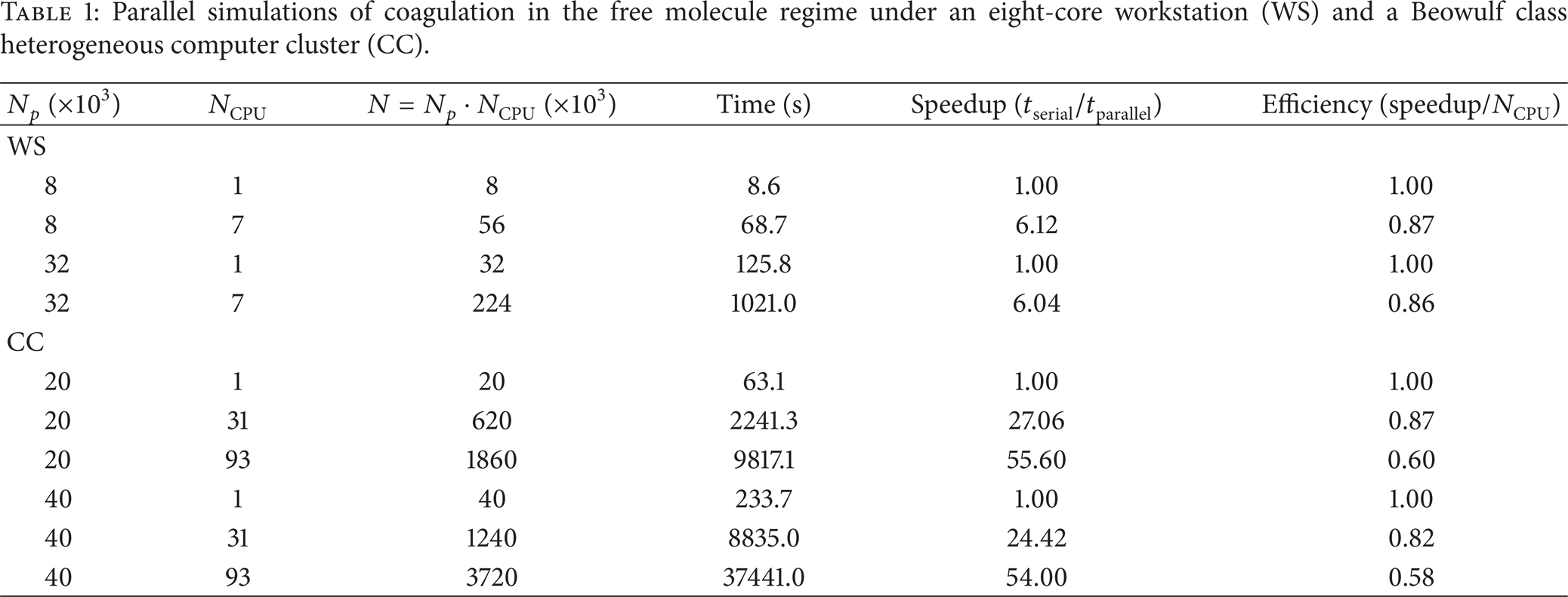
Table 1 compares the computational efficiency with different number of MC particles. First, it is found that the computation time is proportional to N2 (with relative error less than 10%) when using the same number of computing processors, which agrees with the above analysis. This fact is used to estimate the serial computation time tserial with large number of particles, which cannot be handled by a single processor, by scaling the computation time for a small serial simulation. The tserial obtained in this way is then used to calibrate the efficiency of parallel computing. The efficiency is found insensitive to N p (number of particles per processor) with fixed NCPU (number of processors) and only marginal decreasing is observed when increasing N p . While increasing NCPU, the parallel efficiency decreases more significantly. Overall, the parallel efficiency is satisfactory. Under the workstation, it reaches 87% and 86% for the two parallel simulations, respectively. Under the computer cluster, it also reaches 87% for a half million particles simulation. For a 3.7 million simulation, it is 58%. Such efficiencies are promising.
Parallel simulations of coagulation in the free molecule regime under an eight-core workstation (WS) and a Beowulf class heterogeneous computer cluster (CC).
3.2. Free Molecule Regime Coagulation
The setup of free molecule regime coagulation testing case is introduced in Section 3.1, where the efficiency of parallel computation is discussed. In this section, the accuracy will be addressed.
There is no general analytical solution available for the free molecule regime coagulation. However, after introducing the self similarity transformation, the Smoluchowski's equation can be reduced to an ordinary integrodifferential equation, and the normalized PSD will be self-similar after enough long time [1]. Specifically, the total particle number density N∞ is found to satisfy [45]

where the constant a is an integral function (Friedlander [1, (page 261)] gave a = 6.67 by numerical analysis.) of the similarity transformation introduced, ϕ is the volume fraction, and A is a short notation of the corresponding part. Since both T and ϕ are constant for this case, it is easy to integrate the equation to render

Figure 3 compares the number density obtained by the MC simulation, a sectional method [46], and two solutions evaluated according to (9) with the constant a = 5.80 and a = 6.67, respectively. The MC simulation and the sectional method give nearly identical solutions. The analytical solution with fitted constant (a = 5.80) also agrees very well with the numerical solutions. But the solution (a = 6.67) given by the numerical analysis of Friedlander [1] differs slightly from the others. It is worth pointing out that a = 6.67 is obtained after the PSD has reached self similarity state, but within the simulation time tstop = 4.5 ms the PSD is far from self similarity (see discussion below). The simulation shows that a smaller constant a = 5.80 can be used to predict the evolution of the total number density before the PSD reaching the self-similarity state. It is also worth pointing out that the MC simulation uses only 50 particles. This reveals that the evolution of number density is insensitive to the precise form of the PSD, since simulations with N = 50 may not capture the PSD very accurately. This conclusion coincides with that for constant kernel coagulation. Simulations of coagulation in the continuum regime also comes to the same conclusion (results not shown in this paper). Consequently it seems that the MC simulation can always predict the number density very accurately with only a few dozens of particles with enough repetitions to limit the uncertainty.
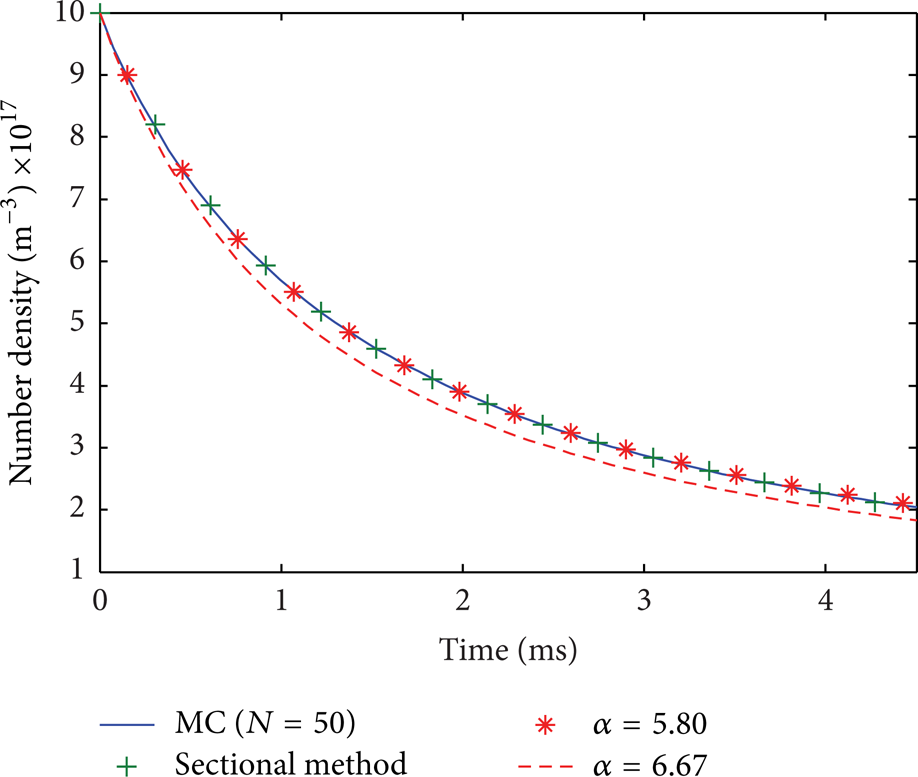
Evolution of the number density for coagulation in the free molecule regime. The program code for the sectional method is from Prakash et al. [46], enough number of bins has been used to render the solution presented here. a = 5.80 and a = 6.67 are the constants in the similarity transformation solution (9) corresponding to numerical fitting (current) and numerical analysis in Friedlander [1].
The self-similar solution of the PSD (after reaching the self-similar form) means that if the PSD at one time is known, then the PSD at any other time is also known through the self-similar transformation. Figure 4 shows the PSDs at two different times. Both times are larger than the time required to reach the self-similar form, tSP, which is estimated as tSP = 0.02's according to [1, Equation (7.78)]. With N = 50, the simulated PSDs exhibit good self-similar property, and also agree with that from a larger simulation of N = 3200. However, it is clear that the simulation with small N cannot correctly predict the number density of large particles (η>10 in Figure 4), which agrees with the findings in the simulations of constant kernel coagulation.

Self-similar PSD (histogram) for coagulation in the free molecule regime. The results are obtained by averaging over 10000 repeated MC simulations. ϕ is the volume fraction. η is dimensionless particle volume normalized by the average volume (ϕ/N∞). ψ is the normalized particle density function.
3.3. Free Molecule Regime Coagulation and Constant Rate Nucleation
This testing case has the same setting as introduced in Section 3.1 for coagulation, and it also includes nucleation with constant rate of 1.0 × 1020 m−3·s−1. Nucleated particles are assumed to have the same diameter 1.5 nm as that of the initial particles.
Figure 5 shows the simulation results with different N. Solutions from the sectional method [46] are also given as reference. All these numerical results, that is, number density, volume fraction and second moment and so forth, agree with each other very well. The second moment given by the sectional method is slightly bigger than those from the MC simulations. It is believed that the sectional method has lower accuracy than the MC simulation with N = 2000. Since increasing N further also gives the same solution as N = 2000. But it is hard to increase the accuracy of the sectional method used here to very high level [46].

Results for free molecule regime coagulation and constant rate nucleation. (a) number density, (b) average diameter, (c) total particle volume, and (d) second moment.
3.4. Condensation and Coagulation
Analytical solutions for condensation and coagulation (Of course, the collision kernels are limited to the three types that the solutions to the Smoluchowski's equation are known.) are available for a few special cases [47]
α(v) = α0v, β(v,u) = β0,
α(v) = α0v, β(v,u) = β0·(v + u),
α(v) = α0, β(v,u) = β0,
where α(v) is the condensation rate, α0 and β0 are constants.
In case (iii) condensation and coagulation are independent with each other, which is a trivial case for the current MC method that intrinsically separates the two processes. Here, results for case (i) from MC simulation are reported. For simplicity, the parameters are set as α0 = 1, β0 = 1. The initial PSD is

which has total number density 1, and average volume 1.
Under these conditions, the solution for case (i) is [47]
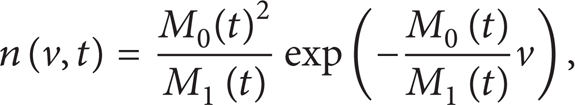
where the zeroth and first moments, M0 and M1, are
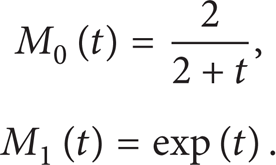
Further, we derive the second and third moments here
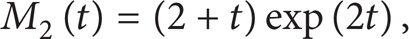
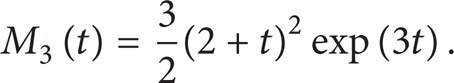
Figure 6 compares the MC simulations (Strang1 scheme) of the condensation and coagulation case with the analytical solutions (12)–(14), which agree extremely well with each other. First order Lie1 and Lie2 schemes are found to give almost the same results as the Strang1 scheme (not shown). In this case, coagulation is independent of condensation, and condensation does not change the number density, hence the solution for the number density is the same as the case of pure constant rate coagulation (not shown here). Since the condensation rate is a linear function of particle volume, the evolution of volume fraction M1 is deterministic, which has increasing rate

Evolution of moments for condensation and coagulation with Strang1 scheme, δt = 0.1. (a) M0, (b) M1, M2 and M3. Strang1 scheme, δt = 0.1.
4. Conclusions
A new hybrid algorithm combining stochastic simulation and deterministic method for solving the PBE for aerosol dynamics is proposed. Nucleation and surface growth are handled with deterministic means, while coagulation is simulated with a stochastic method (Marcus-Lushnikov stochastic process). Operator splitting techniques are used to synthesize the deterministic and stochastic parts in the algorithm. The algorithm is parallelized using the MPI.
Parallel simulation of aerosol coagulation in the free molecule regime shows that the parallel computing efficiency decreases with increasing the number of CPUs. While for fixed number of CPUs, increasing the number of MC particles does not affect the efficiency significantly. Near 60% parallel efficiency is achieved for the maximum testing case with 3.7 million MC particles running on 93 CPUs in parallel.
For verification purpose, the algorithm has been applied to a series of testing cases, that is, free molecule regime coagulation, free molecule regime coagulation + constant rate nucleation, and constant kernel coagulation and condensation. All the simulation results agree very well with known analytical solutions or numerical solutions from a sectional method [46]. Generally, it is found that only small number (hundreds or thousands) of MC particles is necessary to accurately predict the aerosol particle number density, volume fraction, and so forth, that is, low order moments of the PSD function. Accurately predicting the high order moments of the PSD needs to dramatically increase the number MC particles.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Footnotes
Acknowledgment
Kun Zhou would like to thank F. Bisetti for insightful discussion and valuable help. Kun Zhou is partially supported by the National Natural Science Foundation of China (Grant no. 11302110). Zhu He is supported by the National Key Technology R&D Program of Chian (Grant no. 2011BAK06B02).