
Abstract
An atomistic structural mechanics method, which is based on the exclusive use of spring elements, is developed in order to study the effect of imperfections due to atom vacancy on the vibrational characteristics of single-walled carbon nanotubes (SWCNTs). The developed elements simulate the relative translations and rotations between atoms as well as the mass of the atoms. In this way, molecular mechanics theory can be applied directly because the atomic bonds are modeled by using exclusively physical variables such as bond stretching. The method is validated for its predictability comparing with vibration results found in the open literature for pristine nanotubes. Then, it is used for the vibration analysis of defective nanotubes. Imperfections such as one-atom vacancy, two-atom vacancy, and one carbon hexagonal cell vacancy are investigated. Their effect on vibrational behavior is explored for different defect positions, nanotube diameters, and support conditions. According to the obtained results, the fundamental frequency is decreased as the size of imperfection increases, and the percentage reduction in fundamental frequency due to the atomic vacancy defect is more affected for a single-clamped SWCNT than for a double-clamped one.
1. Introduction
There is a wide range of applications in which the vibration characteristics of carbon nanotubes (CNTs) are significant. In applications such as oscillators, charge detectors, field emission devices, vibration sensors, and electromechanical resonators, oscillation frequencies are key properties. This fact drives the research interest in studying and developing accurate theoretical models for the natural frequencies evaluation of CNTs. A comprehensive review on the vibrational behavior of CNTs and their composites is given by Gibson et al. [1]. Specifically, they have presented many of the related theoretical research efforts, which involve continuum, atomistic, and hybrid atomistic/continuum models on this field. Molecular mechanics (MM) methods have been in the forefront of the research on studying the mechanical properties [2, 3] as well as vibrational response [4–6] of nanomaterials. This is due to the fact that MM methods have some distinct advances in comparison with other theoretical techniques such as molecular dynamics (MD), quantum mechanics (QM), and continuum mechanics (CM) methods. In many cases, large molecular systems can be modeled successfully while avoiding the required complex quantum mechanical calculations entirely. Therefore MM methods have lower computational cost in comparison with QM or MD while simultaneously being capable of representing the atomistic structure of a nanomaterial in detail in contrast with CM methods.
Recently, Anuar and Isa [7] have presented a review of quite a number of publications on CNTs and their dynamic properties. The main topics that have been covered in this review were the applications of CNTs, their dynamic characteristics including the modeling and simulation of vibrating CNTs, and finally the vibration modes of CNTs. Applying different gradient elasticity theories including stress, strain, and combined strain/inertia ones, Ansari et al. [8] have investigated the vibration response of SWCNTs establishing the theoretical formulations based upon both the Euler-Bernoulli and the Timoshenko beam theories. To validate the accuracy of their analysis, molecular dynamics simulations were also conducted. Kucuk et al. [9] derived variational principles for multiwalled carbon nanotubes undergoing linear vibrations using the semi-inverse method with the governing equations based on nonlocal Timoshenko beam theory which takes small scale effects and shear deformation into account. Hu et al. [10, 11] have presented a brief review of vibrations of SWCNTs investigated using the nonlocal beam model and nonlocal rod model and MD simulation summarizing the applicability of the nonlocal continuum models based on the MD simulation. Fakhrabadi et al. [12] have developed a molecular mechanics based finite element modeling and using a multilayer perceptron neural network have predicted the fundamental vibration frequencies of the CNTs with different diameters and lengths. Kim et al. [13] have analyzed the chirality and length dependence of SWCNTs by using normal mode analysis based elastic network model in which all interatomic interactions of the given SWCNTs structure are represented by a network of linear spring connections. They have pointed out that there is a critical aspect ratio between diameter and length to determine vibration mode shapes, and it can be empirically formulated as a function of nanotube length and diameter. Chowdhury et al. [14] have investigated the vibrational properties of zigzag and armchair SWCNTs using the molecular mechanics approach based on the universal force field. Their results indicate that the natural frequencies decrease as the aspect ratios increase showing similar trends with results of previous studies for CNTs.
As it has already been observed [15], during the synthesis process of CNTs, topological and vacancy defects are commonly introduced in their structure. As the exact mechanisms of nanotube growth are not much known, vacancy defects, such as missing atoms, are likely to exist within the nanostructure. Considering that the dynamic performance of CNT is significant in many applications, the investigation of such defects and their influence on the CNT vibrational response is a crucial issue. Based on this fact, Joshi et al. [16, 17] have modeled CNTs, by considering them as space frame structures consisting of three-dimensional beams and point masses, in order to find their natural frequencies and study the effects of defect like atomic vacancies in them. Parvaneh et al. [18] have employed a Morse potential for stretching and bending potentials, and a periodic type of bond torsion used for torsion interactions to develop the structural model of CNTs. Then, they have investigated the natural frequencies for various CNT aspect ratios and have predicted the effect of different vacancy and Stone-Wales defects on the natural frequency of zigzag and armchair nanotubes. Ghavamian and Ochsner [19] have obtained, through a finite element approach, natural frequencies of CNTs with randomly scattered defects (Si-doping, carbon vacancy, and perturbation) of different amounts and have investigated the influence of these defects on the vibrational stability of nanotubes. Based on the CM mechanics method, Chen et al. [20] have used three-dimensional finite element models to study the effect of different defects on the vibration of CNTs showing that the length, the diameter, and the defect position are important factors which affect the vibration properties of CNTs.
Despite the fact that there is a variety of studies dealing with the vibrational behavior of pristine CNTs, little has been said about the effect of defects in the vibration response of nanotubes [16–20]. However, in those works different numerical schemes have been utilized, which present some distinct limitations in comparison with the one which will be presented in the following. Specifically, in the studies [16, 17, 20] beam elements have been utilized for representing bonds. However, beam elements fail to remain straight while stretched as chemical bonds do. Furthermore, in a beam element analysis the questionable assumption that the bond angle resistance force existing between two bonds is approximately equal to the bending stiffness of a single beam (representing one bond) may not be avoided. With regard to the studies [18, 19], different force potentials have been utilized in comparison with the present effort. Principally, the main uniqueness of the presence of the present analysis lies in the fact that it is focused on the vibration spectra from which the influence of defect becomes clear. In other words, a new atomic defect detection tool is proposed. In contrast to other investigations, finally, the variety of parametric studies conducted here leads to a clear definition of the problem.
In this paper, a spring-mass structural method is adopted which has been basically developed by the same research group of the present study [21, 22]. The efficiency of this method has been tested for static [21] as well as vibrational problems [22]. However, it should be clarified that in the last study [22] only nondefected, pristine CNT structures have been analyzed. The specific method is utilized here in order to examine the vibration behavior of SWCNTs presenting atomic vacancy defects. The analysis is focused on defects in which one or more neighbor atoms are missing from the atomic lattice rather than on topological defects such as Stone Wales ones. The main reason is that atomic vacancies are usually associated with permanent bond breaking and therefore may be characterized as more critical for the structural integrity of the nanomaterial under consideration. Under severe loadings, vacancy defects are more likely to lead to more extensive imperfections, propagating microcracks and thus local or total failure. The proposed numerical technique uses spring elements of appropriate stiffness parameters, obtained by molecular mechanics calculations in order to approach the interatomic behavior. Additionally, it utilizes lamped masses at atomic positions in order to simulate the inertia effects of the nanostructure. The atomic vacancy imperfection is approached by removing the attached springs and mass corresponding to the position where the carbon atom is missing. Assembling the stiffness and mass matrices, the dynamic equilibrium equation is constructed and its solution reveals the natural frequencies and mode shape of vibration of CNTs. The method is successfully validated using results found in the open literature for pristine nanotubes. The frequency behavior of defective CNTs is investigated with respect to the CNT diameter as well as the size and location of defect. To the authors’ best knowledge, the vibration signature difference between pristine and defective CNTs is evaluated for the first time in the literature. The results indicate that detection of CNT imperfections may be detected by comparing the vibration spectra of defective and nondefective CNTs. Such a comparison may lead to valuable conclusions discussed in the following.
2. Computational Model
The CNT is modeled according to its exact three-dimensional geometry [21]. Specifically, nodes are defined at carbon atoms positions and a concentrated mass equal to carbon atomic nucleus mass (m = 1.9943 × 10−26 kg [22]) is added on them via point mass elements. Then the nodes that correspond to carbon atoms are connected with linear elastic spring elements in order to simulate the interatomic interactions between carbon atoms which, by assuming small strains and adopting the simplest harmonic forms, are described by the following total potential energy [23]:
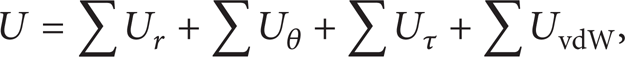
where U r represents the energy due to bond stretching, Uθ the energy due to bond angle bending, and Uτ the energy due to dihedral angle torsion as well as out-of-plane torsion. Finally, UvdW term describes the energy due to van der Waals nonboned interactions, which are negligible for single-walled CNTs [13, 16, 20]. The above energies are given by the following equations [24], correspondingly:
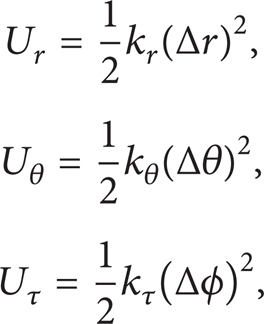
where k r , kθ, and kτ are the bond stretching, bond angle bending, and torsional resistance force constants, respectively, while Δr, Δθ, and Δϕ represent the bond length, bond angle, and twisting bond angle variations, respectively. The values of these parameters are k r = 6.52 × 10−7 N nm−1, kθ = 8.76 × 10−10 N nm rad−2, and kτ = 2.78 × 10−10 N nm rad−2 [23]. The second derivatives of the potential energy terms appearing in (2), with respect to bond length, bond angle, and twisting bond angle variations, respectively, produce the spring stiffness coefficients k r , kθ, and kτ according to Castigliano's theorem. It should be noted that the force constants adopted here are appropriate for normal temperatures. Under different thermal conditions different force constant values and bond lengths should be utilized. However, the analysis of the temperature effect is out of the scope of the present effort.
The spring elements used in the present analysis are essentially two-noded with six degrees of freedom per node, that is, three translations and three rotations. Applying the conventional displacement formulation, their equilibrium equation in their three-dimensional local coordinate system

where the superscript e identifies the particular spring element,
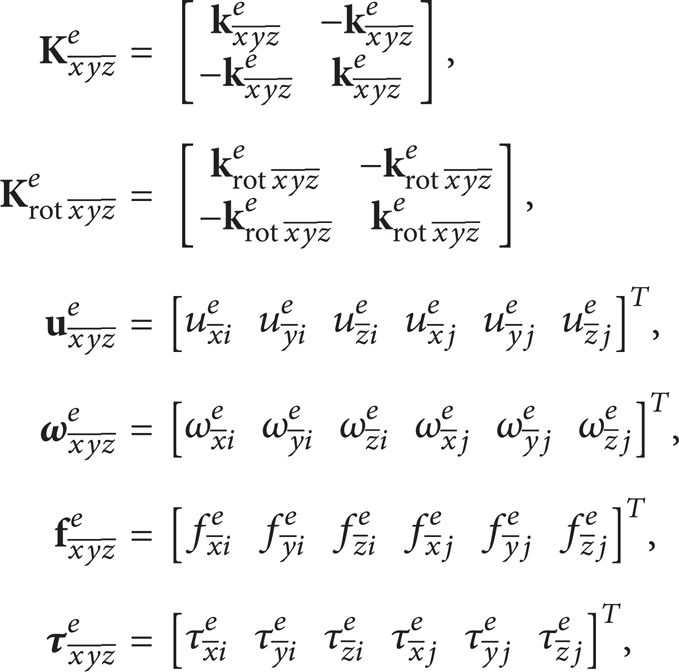
where and i and j are the two nodes of element e and
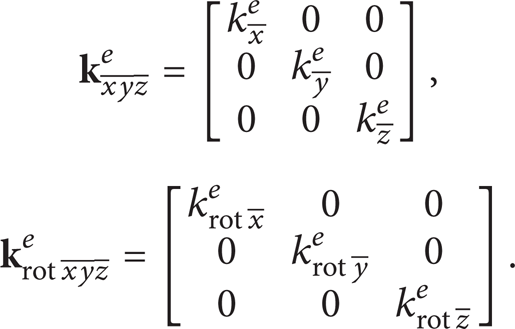
Two types of spring elements, which are depicted in Figure 1, are used in the proposed analysis. The first type of spring element is utilized to represent the bond stretching as well as twisting interatomic interaction and will be called hereafter cc element. The displacement and rotation stiffness matrices of the specific element, respectively, may be expressed as
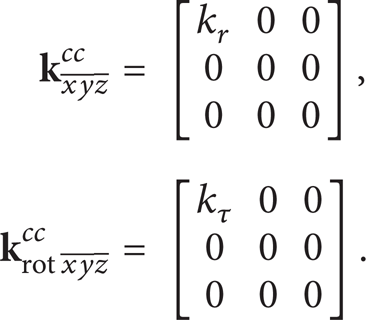

Spring and mass element representation of carbon interatomic interactions.
The second, called hereafter ccc element, simulates angle bond bending interaction. As it may be observed (Figure 1), for simplicity reasons, the angle bending interaction is equivalently represented by the use of a straight spring which interconnects the edge atoms of a c-c-c nanostructure. In this way, the rotation stiffness kθ may be replaced by a displacement stiffness k l . The stiffness k l is a distance dependent parameter which is equal to

where r0 and l0 are the distance between two covalent bonded atoms and the distance between two opposite atoms in a c-c-c nanostructure in the undeformed state, respectively. On this basis, the displacement and rotation stiffness matrices of the ccc element are expressed by the following relationships, respectively:

Finally, a point mass element is utilized at each carbon position (Figure 1). This element, called hereafter c element, is one-noded and has the following elemental equation in the global coordinate system (x, y, z):

where the dot denotes the derivative with respect to time, and

Note that this one-noded element which is independent of the rest two-noded spring elements is attached uniquely at each atomic position and hence has to represent the whole carbon atom mass.
The stiffness matrix

where
3. Results and Discussion
3.1. Numerical Technique Validation
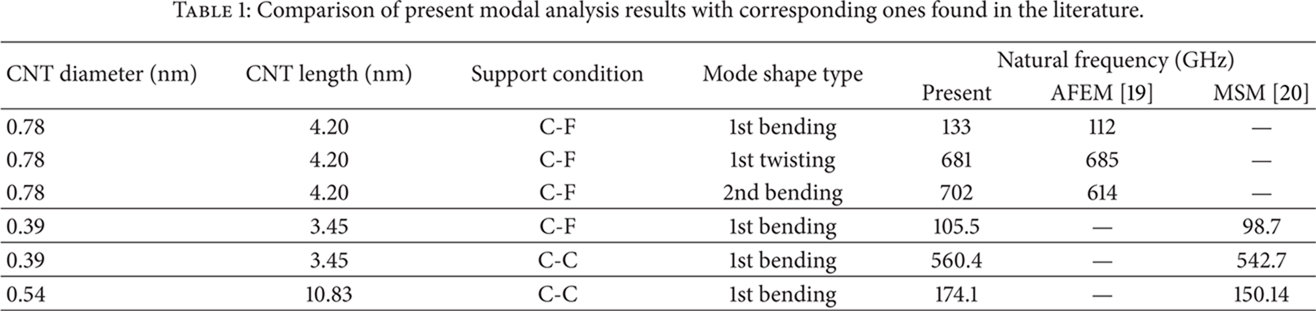
Firstly, in order to validate the proposed method, results obtained by using the proposed formulation are compared in Table 1 with corresponding outputs from other theoretical approaches such as atomic-scale finite element method (AFEM) [25] and beam based molecular structural mechanics (MSM) [26]. The comparison is not focused only on the fundamental frequency which is related to the 1st bending mode shape, but also on higher order natural frequencies corresponding to the 2nd bending mode shape and the 1st twisting mode shape as well [22]. Moreover, various CNT diameters, lengths and different support conditions are tested. Very good agreement is shown between results for all cases. The small discrepancies that may be observed are mainly due to the different numerical formulation method followed. It should be noted that the basic concept of the proposed numerical technique has already been successfully validated for a variety of problems concerning the effective mechanical properties of SWCNTS [21], the vibration behavior of multiwalled CNTs [27], the coupling phenomenon between axial and shear deformations in graphene-based structures [28], and finally the radial stiffness and natural frequencies of fullerenes [29].
Comparison of present modal analysis results with corresponding ones found in the literature.
3.2. Frequency Analysis of Defective CNTs
Using the proposed modal analysis, the natural frequencies of the CNT arise. Specifically, the armchair (5, 5) CNT has been assumed to be clamped-free (C-F) or clamped-clamped (C-C). Four different cases involving structural defection type have been examined: (a) the CNT has been assumed to be perfect, (b) the CNT has been assumed to present a specific point defect, that is, one carbon atom vacancy, (c) it has been assumed that two neighbor atoms are missing from the CNT, and (d) it has been assumed that a whole hexagonal cell is missing from the CNT. The imperfections have been assumed to be positioned at specific locations along nanotube length. The total length of the nanotube L has been taken equal to 11 nm. Figure 2 depicts the geometry of a CNT presenting the three aforementioned types of defection.

Types of defect under consideration in the (5, 5) armchair SWCNT: (a) one atom, (b) two neighbor atoms, and (c) one hexagonal cell vacancy.
Hereafter, the fundamental frequency of a defected nanotube will be symbolized as f while the original fundamental frequency of the corresponding perfect nanotube will be symbolized as f0. Figure 3 illustrates the ratio of the defected CNT fundamental frequency to the perfect CNT fundamental frequency, f/f0, with respect to the nondimensional position of the related defect from the left clamped edge, x/L (Figure 2(a)), and type of imperfection. The larger the imperfection, the higher the drop of natural frequency. This is due to the fact that overall structural stiffness of the CNT is downgraded especially when more carbon atoms are missing. For a given CNT, structural stiffness could be effectively increased by adding supports, reducing bond size, or increasing force constant values. It is evident that near supports enhanced structural stiffness is present. Hence, removing bonds close to this area leads to a significant drop of overall stiffness and hence the decrease of natural frequency. When the support condition is the C-F one (Figure 3(a)), the natural frequency is mostly influenced when the imperfection is placed at the clamped edge of the nanotube. For a given nanotube geometry, the presence of a local defect may lead to a significant fundamental frequency decrease up to 18%. When the support condition is the C-C one (Figure 3(b)), the natural frequency is mostly influenced when the imperfection is placed near the center or the edges of the nanotube. The natural frequency drop observed when the vacancy defects are placed at the center may be explained by the following consideration. The central area of a doubly fixed CNT is far away from supports and thus may be characterized as a high speed vibrating area which has a significant inertia effect. Hence, removing atomic masses close to this area leads to a significant drop of overall inertia and hence the decrease of natural frequency. Here, the reduction in fundamental frequency reaches 7.4% when the defect is located at the middle of the nanotube length and 7% when the imperfection is placed near the clamped edges of the CNT. It is noticed that fundamental frequency reduction is less significant for the C-C case since the overconstraint of the nanostructure conceals the impact of the defect.

The effect of various defects in the (5, 5) SWCNT, which are located at various intermediate positions along its length, on its fundamental frequency for the (a) C-F and the (b) C-C support condition.
Figure 4 depicts how the fundamental frequency of CNTs is affected by a hexagonal cell vacancy located near to left edge of the CNT with respect to its diameter. The support conditions assumed are C-F (Figure 4(a)) and C-C (Figure 4(b)). It can be observed that the fundamental frequency of the defective CNT is lower than the pristine one for all cases. Evidently, the difference between them is slightly reduced as the diameter becomes higher. However, the percentage reduction of fundamental frequency is significantly decreased for higher diameters, due to the fact that the fundamental frequency is increased as the CNT diameter increases.

Fundamental frequency of perfect and defective CNTs versus their diameter for the (a) C-F and the (b) C-C support condition.
3.3. Vibration Signature of Defective CNTs
Figures 5(a) and 5(b) depict displacement spectra concerning the (5, 5) SWCNTs for the C-F and C-C case, respectively. The spectra corresponding to the three types of defect are presented in contrast to the perfect CNT spectrum. In all cases considered, the imperfection is placed near the clamped edge of the singly clamped CNT or near the middle of the doubly clamped CNT in order to investigate the maximum influence effects on vibration response. Generally the peaks of excitations correspond to natural frequencies and obviously the first appearing peak corresponds to the fundamental one. The backward shift as well as the raise of the peaks as the size of defect is increased becomes obvious. Figures 6(a) and 6(b) present displacement spectra for the C-F and C-C CNT case, respectively, involving several positions of one cell vacancy defect. For all cases, the increase of displacement as well as movement of its maximum values along frequency axis is evident.

Vibration spectrum of the (5, 5) SWCNT which is (a) singly clamped with a defect at x/L = 0.09 and (b) doubly clamped with a defect at x/L = 0.45.

Vibration spectrum of the (5, 5) SWCNT, presenting a cell vacancy defect at various positions along its length, which is (a) singly clamped and (b) doubly clamped.
4. Conclusions
A numerical investigation concerning the change in vibration response of defective SWCNTs has been performed. In order to achieve this, a computationally simple and efficient numerical scheme has been developed, for the free vibration analysis of the SWCNTs. The numerical scheme is based on the representation of the atomistic structure of the CNT by the use of three-dimensional spring elements and point mass elements, defined by appropriate stiffness and mass matrices, respectively. The analysis has been conducted in a parametric manner regarding the type of defect, support conditions, and CNT diameter. Using the proposed method several conclusions have arisen.
CNT defect may be detected by the revealed fundamental frequency change.
Even one carbon vacancy may lead to significant fundamental frequency drop.
CNT vibration behavior is more significantly influenced when the imperfection is placed at the support edges.
A middle positioned defect has also a maximum effect for a C-C CNT.
The higher the diameter, the lower the percentage drops in the fundamental frequency provoked by the defect.
The maximum possible percentage reduction in fundamental frequency due to a vacancy defect is higher for the C-F than the C-C case.
The peaks in the vibrational spectra of a CNT get higher values and move backwards along frequency axis as the defect increases in size.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.