
Abstract
Ion channels are critical for neuronal excitability and provide important targets for anticonvulsant drugs. In the past few years, several monogenetic epilepsies have been linked to mutations in genes encoding either voltage-gated or ligand-gated channels. The recognition that certain epilepsy syndromes are “channelopathies” initiates a new era in understanding the molecular pathophysiology of seizure disorders. This review summarizes recent advances related to this exciting area of investigation.
Introduction
Complex genetic factors contribute to the pathogenesis of most types of idiopathic epilepsy, but several recognized epilepsy syndromes are transmitted by mendelian inheritance (i.e., caused by mutations in single genes). Mutations in 11 different genes encoding ion channels account for the majority of monogenic epilepsies (see Table 1 and Fig. 1). Therefore epilepsy joins other paroxysmal disorders of the nervous system, including periodic paralysis, episodic ataxia, and migraine as “channelopathies.” This review focuses on recent work that defines the role of inherited ion-channel dysfunction in the molecular basis of epilepsy.
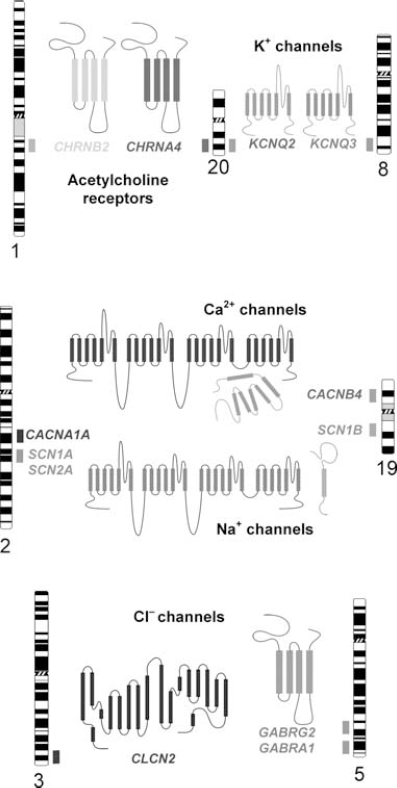
Chromosomal locations of ion channel genes associated with inherited epilepsy. The position of each gene is represented by a shaded box adjacent to the respective chromosome ideogram. A structural model of the corresponding ion channel protein encoded by each gene is also illustrated.
Inherited Epilepsy Syndromes Caused by Ion Channel Mutations

Online Mendelian Inheritance in Man database (http://www.ncbi.nlm.nih.gov/Omim/)
Cytogenetic location defined by using
Ion channels may be broadly classified as either voltage-gated or ligand-gated, depending on whether the primary stimulus for their activity is a change in local membrane potential or a chemical messenger (e.g., neurotransmitter). The role of ion channels in neuronal excitability is well established, and the identification of mutations in neuronal ion-channel genes linked to inherited epilepsy has put emphasis on the delicate balances that maintain electrical harmony in the central nervous system (CNS).
Table 1 lists the currently known inherited epilepsy syndromes associated with mutations in either voltage- or ligand-gated ion channels. Genetic heterogeneity (i.e., the same clinical syndrome caused by mutations in different genes) is evident among these disorders. For example, generalized epilepsy with febrile seizures plus (GEFS+) has been associated with mutations in three voltage-gated sodium channel genes (SCN1B, SCN1A, SCN2A) or the gene encoding the γ2 subunit of γ-aminobutyric acid (GABA)A receptors (GABRG2). The following discussion of specific inherited epilepsy syndromes is organized according to the type of ion-channel gene involved.
Epilepsy Associated with Voltage-gated K+ Channels
Several distinct types of potassium channels are expressed in the nervous system and serve a wide range of functions. They are most critical for maintaining resting membrane potentials and enabling rapid repolarization after action potentials. The syndrome of benign familial neonatal convulsions (BFNCs), a rare inherited form of idiopathic generalized epilepsy (IGE) exhibiting autosomal dominant inheritance, was the first seizure disorder associated with neuronal potassium channel mutations. BFNC is characterized by convulsions occurring in the neonatal period that typically resolve spontaneously after a few weeks of life. In very rare cases, adulthood epilepsy occurs. BFNC is genetically heterogeneous, with two identified loci on chromosomes 8q and 20q. In 1998, the potassium channel gene KCNQ2 was identified as the 20q gene, and a closely related gene, KCNQ3, was discovered to be responsible for the chromosome 8q-linked syndrome (1,2).
In the nervous system, KCNQ2 and KCNQ3 gene products assemble to form potassium channels that generate ionic currents resembling neuronal M-currents (3). M-currents modulate neuronal excitability by dampening the tendency for repetitive firing. Neuronal M-currents are inhibited by muscarinic acetylcholine receptor agonists as well as activators of other types of neurotransmitter receptors. Mutations in either KCNQ2 or KCNQ3 reduce function of the encoded potassium channel by a dominant-negative mechanism consistent with the autosomal dominant inheritance pattern of BFNCs (4).
Epilepsy Associated with Voltage-gated Na+ Channels
Voltage-gated sodium channels are responsible for the rapid membrane depolarization that characterizes the initial “upstroke” of the neuronal action potential. Many conventional anticonvulsants (AEDs) act by blocking sodium channels in a use-dependent manner. Mutations in three voltage-gated sodium channel genes have been discovered in patients affected by several inherited epilepsy syndromes that exhibit febrile seizures. In 1997, Scheffer and Berkovic described GEFS+, a newly recognized epilepsy syndrome with autosomal dominant inheritance (5). The syndrome was named in reference to the common occurrence of febrile seizures in early childhood that often persisted after age 6 years. In addition to febrile seizures, affected adult members of GEFS+ families exhibited afebrile seizures and seizures with multiple clinical phenotypes. Missense mutations in SCN1A encoding a neuronal voltage-gated sodium channel α-subunit account for the majority of GEFS+ cases (6–10), but heritable defects in two other sodium channel genes (SCN1B, SCN2A) (11,12) and a GABA-receptor subunit (GABRG2; see section entitled Epilepsies Associated with GABAA Receptors) (13,14) also can cause the same disorder or clinically similar conditions.
The functional properties of mutant neuronal sodium channels associated with inherited epilepsy have been described (15–18). Three SCN1A mutations associated with GEFS exhibit defects in fast inactivation gating, characterized by a persistent, noninactivating current during membrane depolarization (16). These findings suggest that, in some cases, SCN1A mutations promote a gain-of-function in sodium channels, leading to neuronal hyperexcitability. Other SCN1A mutations associated with GEFS cause other types of functional impairments that may lead to loss of function (15). Whether gain of sodium channel function in excitatory neurons or loss of function in inhibitory neurons is the primary mechanism responsible for epilepsy in GEFS is under investigation.
Severe myoclonic epilepsy of infancy (SMEI) is a rare convulsive disorder characterized by febrile seizures, with onset during the first year of life, which is followed by intractable epilepsy, impaired psychomotor development, and ataxia (19,20). Seizures in this disorder are usually unresponsive to AEDs. Recently, >60 heterozygous SCN1A mutations, many of which are de novo mutations, have been reported in SMEI, including missense, nonsense, and insertion/deletion alleles (21–24). The observed clinical similarities between SMEI and GEFS, including the frequent occurrence of febrile seizures and shared molecular genetic etiology, lend support to the theory that the two disorders represent a spectrum of severity of the same disease (25). Many SCN1A mutations associated with this disorder appear to encode nonfunctional sodium channels, leading to the suggestion that SMEI is caused by loss-of-function mutations.
Epilepsy Associated with Nicotinic Acetylcholine-Receptor Subunits
Neuronal nicotinic acetylcholine receptors (nAChRs) are located in presynaptic membranes of the cerebral cortex, where they facilitate both excitatory and inhibitory neurotransmitter release. These receptors are pentameric complexes with variable subunit composition. The most common nAChR composition contains α4 and β2 subunits, and mutations in genes (CHRNA4, CHRNB2) encoding these distinct nAChRs have been associated with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (26,27). Individuals affected with ADNFLE experience short, partial seizures during sleep. Episodes often occur in clusters, localize to the frontal lobes by ictal EEG recordings, and involve nonspecific auras along with brief motor seizures. Most cases are mild and respond well to carbamazepine treatment.
Functional characterization of mutant nAChR subunits have suggested either a loss or gain of receptor function (28), making a precise molecular mechanism underlying this epilepsy syndrome difficult to determine. A recent study revealed that mutations in both α4 and β2 nAChR subunits interfere with Ca2+ modulation of receptor function in a dominant manner, suggesting an alternative explanation for ADNFLE (29). Acetylcholine activation of wild-type α4β2 receptors is normally potentiated by extracellular Ca2+. In rapidly firing excitatory synapses, postsynaptic glutamate receptors deplete local extracellular Ca2+ and reduce Ca2+ potentiation of nAChRs, a possible negative-feedback mechanism to limit further presynaptic glutamate release. This feedback mechanism may be disabled in presynaptic membranes expressing mutant α4β2 receptors, potentially leading to increased excitatory neurotransmitter release under some conditions, such as rapid, synchronous neuronal firing during sleep (29).
Epilepsy Associated with Calcium Channels
Voltage-gated calcium channels, particularly P/Q type channels, are important for neurotransmitter release in the CNS. At least three inherited, paroxysmal neurologic syndromes, including episodic ataxia, familial hemiplegic migraine, and spinocerebellar ataxia, have been linked to mutations in a neuronal calcium channel α-subunit gene (CACNA1A). Mutations in three voltage-gated calcium channel genes in mice (Cacna1a, Cacnb4, Cacng2) have been associated with generalized cortical spike–wave discharges, which are potential animal models of human absence epilepsy (30). Despite their strong appeal as candidate genes for human epilepsy, calcium channel mutations have rarely been observed in seizure disorders. One premature stop codon (nonsense) mutation in CACNB4 was identified in a small family affected by juvenile myoclonic epilepsy (31). This gene encodes the β4-subunit, a protein that modulates function and trafficking of P/Q-type neuronal calcium channels. A de novo heterozygous nonsense mutation in CACNA1A also has been reported in a young male patient affected with generalized epilepsy (tonic–clonic and absence seizures) and paroxysmal ataxia (32). Both CACNB4 and CACNA1A mutations cause reduced calcium channel function (31,32). In populations with epilepsy, evidence for an association between the CACNA1A locus and susceptibility to IGE was reported by one group (33) but not confirmed by an independent study (34).
Epilepsies Associated with GabaA Receptors
Type A GABA receptors (GABAA) are ligand-gated chloride channels that mediate inhibitory neural activity. In the healthy postnatal brain, activation of GABAA receptors triggers an influx of chloride ions, which renders the postsynaptic membrane potential more negative (hyperpolarization). This hyperpolarization counters excitatory synaptic inputs that would otherwise depolarize the postsynaptic membrane and promote action-potential firing. The ability of GABAA receptors to mediate chloride influx is dependent on other factors, including potassium–chloride co-transporters and voltage-gated chloride channels that maintain a low intracellular chloride concentration.
Mutations in two genes that encode subunits of GABAA receptors and a third gene encoding a voltage-gated chloride channel have been associated with inherited epilepsy. GABAA receptors are composed of five subunits encoded by multiple different gene families (α, β, γ, δ, ∊, π, θ), with a predominance of complexes containing combinations of αβγ or βδ. The gene encoding the α1 (GABRA1) subunit has been linked to an autosomal dominant form of juvenile myoclonic epilepsy. Mutations in GABRG2 encoding the Γ2 subunit have been associated with GEFS and the syndrome of childhood absence epilepsy with febrile seizures (13,35). Experiments to characterize the function of mutant GABAA receptor subunits have demonstrated impaired receptor activity in vitro suggesting reduced GABA-mediated synaptic inhibition as a primary cause for neuronal hyperexcitability (36).
Epilepsies Associated with a Voltage-gated Chloride Channel
Another gene encoding a chloride-transporting protein associated with inherited epilepsy is CLCN2. CLCN2 encodes a chloride channel that is widely distributed in the nervous system and has a suspected role in neuronal excitability. Mutations in CLCN2 have been associated with IGE in three families exhibiting autosomal dominant inheritance and multiple seizure phenotypes, including juvenile myoclonic epilepsy, juvenile absence epilepsy, childhood absence epilepsy, and epilepsy with grand mal seizures on awakening (37). CLCN2 was originally suspected based on genetic linkage data indicating that a region near chromosome 3q26 was a susceptibility locus for IGE, along with its suspected role in neuronal excitability. Two CLCN2 mutations completely abolish chloride channel function. However, a third mutation (G715E) enables chloride channel activation at less negative membrane potentials. Therefore distinct cellular mechanisms may account for epilepsy associated with different CLCN2 mutations.
Summary
Ion channels are critical for normal neuronal excitability, and heritable defects in these molecules account for many forms of inherited epilepsy. By studying the molecular basis of these rare monogenic epilepsy syndromes, it may be possible to infer molecular mechanisms of epileptogenesis relevant to more common and genetically complex forms of the disease. Similarly, by studying the physiologic impact of specific mutations, we discover the diversity of cellular mechanisms that can promote neuronal hyperexcitability and learn more about the importance of ion channel genes expressed in the brain. These advances also contribute to our ability to recognize and diagnose inherited neurologic diseases, provide important information that is useful for counseling affected families, and identify potential new targets for anticonvulsant therapy.
Footnotes
Acknowledgment
Dr. George is supported by a Javits Neuroscience Investigator Award from the National Institute of Neurological Disorders and Stroke (grant NS32387).