
Abstract
It is a central tenet of the epilepsy field that seizures result from the imbalance of excitation over inhibition (1). The bulk of excitation is mediated by the neurotransmitter glutamate, whereas inhibition results mainly from the actions of γ-aminobutyric acid (GABA). In the neocortex and hippocampus, the intrinsic sources of GABA are the interneurons, which lately have come under intense scrutiny. It has become clear that a large number of distinct types of interneurons can be differentiated in part by the array of neuropeptides they coexpress (cf. 2). Evidence is emerging that the neuropeptide complement of interneurons plays important roles in the way that interneurons regulate excitability. Here we discuss what is known about the relation of one well-characterized neuropeptide, neuropeptide Y (NPY), and epilepsy in experimental animals and humans, and suggest possible roles for the receptors as targets for the control of excessive excitation in epilepsy.
Early investigations focused on the actions of NPY in the hippocampus. We (5–7,) and others (8) observed that application of NPY to freshly prepared slices of rat hippocampus maintained in vitro potently and selectively reduced synaptic excitation mediated by glutamate release (Fig. 1A). Further work showed that the action of NPY was highly selective, inhibiting only excitatory inputs onto pyramidal cell throughout Ammons horn in the rat, but not affecting either synaptic inhibition (7,8) or inputs to dentate granule cells (9), despite the very high levels of NPY in the molecular layer of the dentate (10). The receptor or receptors involved in these actions belong to the G protein–coupled superfamily (11; see later).
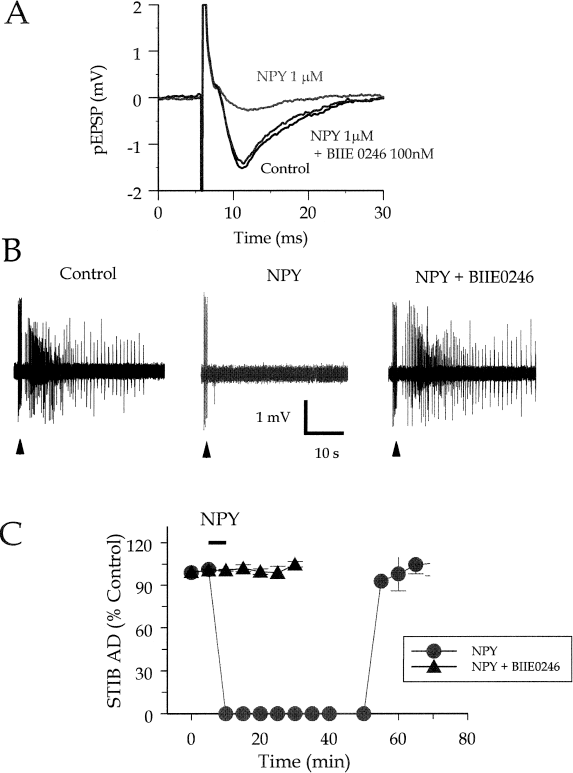
Actions of neuropeptide Y (NPY) and a Y2 antagonist in rat hippocampal slices. A: effect of npy on the population excitatory postsynaptic potential (pEPSP) evoked by electrical stimulation of stratum radiatum in area CA2, recorded extracellularly in area CA1. Black trace, control response; red trace, response in the presence of 1 mM NPY; blue trace, response to NPY in the presence of the Y2-receptor antagonist, BIIE 0246 (100 nm). B: Effect of npy on primary afterdischarges (18 ADs) in the stimulus train–induced bursting (STIB) model in rat hippocampal slice. Extracellular recording from area CA3 shows tonic–clonic 18 AD in control (left trace) in the presence of NPY (300 nm) (center trace), and in 300 nm npy after pretreatment with BIIE 0246 (100 nM). C: time course of the inhibition of the 18 AD by NPY (300 nm) in the absence (red circles) and presence (blue triangles) of BIIE 0246 (100 nM). (B and C modified from ref. 53, with permission).
Detailed studies of the action of NPY were consistent with an entirely presynaptic site in rat hippocampus. Specifically, in neurons whose glutamatergic inputs were suppressed by NPY, the peptide did not affect the postsynaptic responses to glutamate application (6,12). Consistent with this, the release of glutamate from hippocampal slices was shown to be suppressed by NPY (13). Further studies showed that NPY suppressed N-type Ca2+ currents at presynaptic terminals in neuronal cultures (14) and N-, P/Q-, and other types of Ca2+ currents in presynaptic terminals of freshly prepared hippocampal slices (15). Once the sites and mechanisms of action were clear, the next question was what role NPY actually played in the physiology and pathophysiology of the hippocampus.
It appears that elevated activity in hippocampal circuitry, such as that accompanying epileptiform discharges, results in increased NPY expression in interneurons and dentate granule cells, but that prolonged overstimulation in some epilepsy models and in humans ablates NPY (and other) interneurons. Thus several laboratories determined that NPY peptide and messenger RNA (mRNA) expression was increased in epilepsy models in vivo (16–18). At about the same time, it was reported that NPY/GABA interneurons in the hippocampus were selectively ablated in rat epilepsy models in vivo (19,20) and in epilepsy patients (21), although it was recently questioned whether NPY cells are indeed selectively vulnerable in human epilepsy (22). Further work suggested that NPY is expressed ectopically in dentate granule cells of the rat after increased activity (23–25), although this does not appear to be the case after prolonged chronic activity (22,25). However, the increased expression appears to be genuine, because in hippocampal slices prepared from fully kindled rats, the release of NPY is greatly increased over that of controls (26). Whereas some of this release arises from NPY interneurons, the majority is most likely to arise from the massive expression of NPY in the mossy fibers (26). Thus work by several laboratories suggested that NPY might be involved in the hippocampal response to epileptogenesis.
Coupled with the observations of inhibitory NPY actions on glutamate release, the hypothesis emerged that NPY normally controlled excitability in the hippocampus. This was tested in the following ways.
Epilepsy Models in Vitro
Several models mimic features of the acute pathophysiology of epilepsy in vitro. Interictal bursting caused by removal of extracellular Mg2+ (27–29) or by blockade of GABAA receptors with picrotoxin (27,29) are sensitive to actions of NPY. Similarly, in the in vitro models of ictal activity such as the stimulus train–induced bursting (STIB; 30) and related models (31), the primary afterdischarge mimics the tonic–clonic characteristics of limbic seizures and responds to clinically effective concentrations of anticonvulsants (AEDs) that are effective on complex partial seizures (32). NPY, peptide YY (PYY), and the Y2 receptor–selective agonist, [ahx5–24]NPY, potently inhibited the afterdischarge (29; see later for details about NPY receptors), whereas the Y1 and Y5 receptor–preferring ligand, Leu31, Pro34NPY was without effect on the afterdischarge in this model (29,33), suggesting the importance of the Y2 receptor in NPY action (see later). Finally, NPY has been tested in slices of the frontal neocortex against seizure activity induced by a 0-Mg2+ solution. In this preparation, NPY also suppressed seizure activity, but the results differed from those in hippocampus in two ways: first, that it appeared to be a Y1/Y5 receptor mediating the effect; and second, that the duration and frequency of individual interictaform bursts were reduced (34,35). The reason for these differences in the actions of NPY in the hippocampus is unclear. However, overall, the actions of NPY in hippocampal epilepsy models were consistent with a presynaptic site of action.
Epilepsy Models in Vivo
In several models of epilepsy in vivo, the anti-epileptic effects of NPY have been studied. When rat brain GABAA receptors are blocked in vivo with picrotoxin, NPY injection into hippocampal area CA1 inhibited the epileptic behavior induced by picrotoxin pretreatment (36). In a kainic acid model in vivo, NPY injection into the lateral ventricle suppresses motor seizures activity and greatly shortens the kainate-induced ictal EEG recorded in dentate gyrus and CA3 (37,38). Similar results were observed with seizures induced by hippocampal rapid kindling (37,38). The prolonged infusion of NPY into the hippocampus of classically kindled rats also delayed the progression of hippocampal kindling, whereas anti-NPY antibodies applied the same way accelerated the progress of kindling (39).
NPY application in vivo also reduces seizure susceptibility in mice (40,41). Thus in vivo results in several different models of epilepsy or, really, epileptogenesis, are consistent with a powerful antiexcitatory, antiepileptogenic action of NPY.
NPY Receptors and Antiepileptic Actions
Several receptors for NPY have been identified and cloned, the most prominent of which in the hippocampus and neocortex are the Y1, Y2, and Y5 (11). Considerable effort has gone into identifying the receptor or receptors involved in the antiepileptic actions of NPY. In vitro, early work with fragments of NPY suggested that the pharmacologic profile of the presynaptic actions of NPY in hippocampal area CA1 fit that of the Y2 receptor (42). However, it became clear as more receptors were discovered and characterized that these agonist fragments were not adequately specific. Nevertheless, results from in vivo work with the first available antagonist, the Y1 antagonist BIBP 3226, were intriguing, as they suggested that the activation of the Y1 receptor was proconvulsant, and blockade of Y1 receptors was anticonvulsant (43). Unfortunately, different antagonists needed for the unambiguous testing of the inhibitory role of NPY in the regulation of excitability either have not been available or their physical properties make them not amenable to in vivo experiments. The role of endogenous NPY in the regulation of excitability in vivo has therefore been explored by using alternate approaches.
Genetic Manipulation of NPY and Receptor Expression
A rat strain made to overexpress NPY transgenically showed a significant reduction in the number and duration of EEG seizures induced by kainic acid or electrical kindling and exhibited a decrease in the susceptibility to epileptogenesis, correlated with a strong expression of NPY mRNA in the hippocampus (44). Thus an increase in the normal expression of NPY results in a decreased susceptibility to seizures.
Consistent with this, the absence of NPY in mice increases their susceptibility to seizures. Thus in mice in which the NPY gene has been genetically ablated, mild, spontaneous seizures were reported, and their seizure thresholds to PTZ were reduced (41). In another study with these NPY knockout animals, latency and threshold for kainate-mediated seizure induction was identical with that of wild-type littermates, but the NPY knockouts progressed to an uncontrollable status epilepticus and died if not pretreated with NPY (40). Thus evidence from these experiments suggests that endogenous NPY plays a critical role in regulation of excitability. However, these experiments did little to address the nature of the receptor(s) involved.
Mice lacking the Y5 receptor were tested for seizure threshold and susceptibility (45). Y5-receptor knockout mice do not exhibit spontaneous epileptic activity; however, they revealed an enhanced sensitivity to kainic acid–induced seizures (45). Furthermore, slices prepared from Y5 knockout animals were insensitive to the actions of NPY (45,46). Based on these results, it appeared that the Y5 receptor was the dominant receptor in antiepileptic actions of NPY. A further interesting observation was that the Y5 knockout mice were less susceptible to death in response to kainate injections. As an aside, the lethality of seizures may partly be related to the background genetic strain of the mice carrying the knockout, as Y5-receptor knockouts in an inbred SV129 line were much more susceptible to status epilepticus than were Y5 knockout animals bred to a mixed C57BL/6J × SV129 background (45).
These results were somewhat surprising, based on the high levels of Y2 relative to Y5 receptors in the hippocampus (47), and the significant induction of Y2 receptor mRNA and expression (48). Although there is certainly evidence for presynaptic Y5 receptors in the hippocampus, there is no evidence that these receptors are capable of suppressing limbic seizure activity in the STIB model (49).
Our laboratory recently tested this question by using a newly developed antagonist to the Y2 receptor, BIIE 0246 (50), which profoundly reduced the presynaptic response to NPY and to the selective Y2-receptor agonist, [ahx5–24]NPY (51). Indeed, 30 nM BIIE 0246 reduced the effect of NPY in area CA1 of hippocampal slices to 40% of control levels (52; Fig. 1A). The antagonism did not appear to be competitive in hippocampal slices, but the antagonist does behave competitively on Y2 receptors on membranes (50,53), so the apparent noncompetitive action could be attributed to the lipophilic properties of the antagonist (52). Most important, in STIB preparations in which NPY potently suppresses epileptiform activity, BIIE 0246 completely and potently blocks the effect of NPY (52; Fig. 1B and C). Furthermore, a potent and highly selective Y5 agonist was entirely without effect on the epileptiform activity in the STIB model, either alone or in the presence of BIIE 0246 (52). More recently, our laboratory has begun experiments on mice lacking the Y2 receptor. Preliminary results suggest that neither NPY, Y2 agonists, nor Y5 agonists have any effect either on the population excitatory postsynaptic potential (pEPSP) or the epileptiform discharge in the STIB model in Y2 knockout animals (54).
Clearly NPY can have potent actions in models of epilepsy, but the ultimate test of its relevance to epilepsy is in the human. We (55,56) and others (57) examined the NPY effects on neurons from epilepsy patients. NPY had a potent and prolonged presynaptic inhibitory effect on excitatory synaptic transmission in human dentate gyrus, similar to the actions reported in Ammon's horn of the rat. The pharmacology of NPY receptors in the human may be more complex than that in the rat, but preliminary evidence suggests the presence at least of Y2 receptors (56). It would be of considerable interest for potential theraputic applications to determine whether the NPY receptors in human dentate gyrus are on sprouted mossy fibers, or whether they are on conventional inputs to dentate granule cells.
Overall, then, NPY and its receptors have intriguing properties for an endogenous anticonvulsant system in the hippocampus. Unlike that of many inhibitory transmitters, the action of NPY is restricted to presynaptic sites in Ammon's horn and is specific to terminals releasing excitatory amino acids onto principal neurons in the hippocampus. The plasticity of the NPY system in response to elevated activity can generally be considered adaptive. Thus NPY expression and that of the Y2 receptor are upregulated with elevated activity, whereas expression of the proconvulsant Y1 receptor decreases. Furthermore, the ectopic expression of NPY in dentate granule cells, after even mild elevations in activity, can be considered adaptive, as the mossy fibers have a very sensitive presynaptic response to NPY. NPY also acts in epileptic human brain tissue, arguably the ultimate pharmacologic target in this field of research.
However, a number of very important questions remain. Which receptors are the most important in the antiepileptic actions of NPY? How can the conflicting data regarding the receptors be reconciled? What does NPY do in the neocortex and other areas related to seizure generation and spread? Most important from a therapeutic standpoint, can a selective, preferably nonpeptide agonist be generated that will activate the correct receptor in the CNS? These and many other questions are being actively studied, and interesting answers will continue to emerge.
Footnotes
Acknowledgments
We thank the Canadian Institutes for Health Research, The University of Alberta Hospitals Foundation, and the Human Frontiers Science Program Organization. W.F.C. is a Medical Scientist of the Alberta Heritage Foundation for Medical Research.