
Abstract
Landau-Kleffner syndrome (LKS), or acquired epileptiform aphasia, is an epilepsy syndrome involving progressive neuropsychological impairment related to the appearance of paroxysmal electroencephalograph (EEG) activity. LKS appears to share a common pathophysiologic mechanism with continuous spike-wave of sleep (CSWS), acquired epileptic opercular syndrome (AEOS), and even benign childhood epilepsy with centrotemporal spikes (BECTS), with differentiating factors including age of onset, area of primary epileptogenicity, and severity of clinical presentation. This article covers the clinical, diagnostic, therapeutic, and prognostic features of LKS. In a child with autistic spectrum disorder, the presence of a fluctuating clinical course or regression should raise suspicion for the presence of associated epilepsy.
Introduction
Clinical Manifestations
Landau-Kleffner syndrome is characterized by acquired aphasia and paroxysmal, sleep-activated EEG paroxysms predominating over the temporal or parieto-occipital regions. Secondary symptoms include psychomotor or behavioral disturbances and epilepsy with a favorable outcome for seizure control. The prevalence is unclear. A male predominance exists, with an approximately 2:1 ratio. This regressive syndrome affects children after having achieved early developmental milestones, with 3–9 years being the usual age of presentation (4).
The first manifestation of the language disturbance is an apparent “word deafness,” or auditory verbal agnosia (5). Parents report a child no longer responds to their commands, even with raised voices. This auditory agnosia extends to familiar noises including bells, whistles, or a ringing phone. Audiograms and brainstem auditory evoked response (BAER) are normal. Delays and abnormalities in long-latency cortical evoked responses suggest localization to the posterior temporal regions (6). Dichotic listening tasks have shown permanent one-ear extinction contralateral to the affected temporal cortex, and a study of long-latency auditory evoked potentials in five children having recovered from LKS revealed unilateral voltage reduction involving the N1c peak, arising from associative auditory areas, versus a normal N1b peak related to primary auditory cortex (7). The findings suggest long term dysfunction of associative auditory cortex.
Word deafness can deteriorate into total unresponsiveness and impaired expressive communication. Expression is marked by a gradual increase in misarticulations and telegraphic speech; a fluent jargon, or total mutism can occur (5,7). The children may express themselves with a crude sign system or gestures (5). The language disorder can be progressive or incremental, characterized by remissions and exacerbations. The fluctuating course of aphasia in LKS remains among its most puzzling features.
A relationship between younger age of onset and worse longterm outcome has been reported. Bishop (4), in an analysis of 119 reported cases up to 1985, argued that the association between age of onset and prognosis suggests LKS is a disorder of higher-level auditory processing. In a younger child without advanced language development, the effect is devastating, as the normal auditory route leading to acquisition of language is blocked. In the older child the result will be less severe, since language has been partially learned. An analogous scenario is profound deafness, when Wernicke's area is intact but the sensory input is defective. Profound deafness in a young child has a more profound effect on language development than in an older child. Even the relatively mild and fluctuating hearing loss of otitis media may impair language in a young child in the process of developing language (8). Children with LKS may not follow precisely an auditory processing disorder model (9). Given the usual age of presentation between 4 and 7 years, the child has not acquired sufficient reading and writing skills; the older child, however, may lose these skills. Partial retention of writing skills suggests a better prognosis in the reeducation phase (10).
The language disorder of LKS has commonalities with autism spectrum disorder. Communication deficits in autism include abnormal development of spoken language and impaired ability to initiate or sustain conversation. The autistic child's language is often stereotyped, repetitive, and idiosyncratic, with echolalia and neologisms (11). Confusing the picture is the fact that seizures may occur in autism, and EEG abnormalities are common (12). Furthermore, at least a third of autistic toddlers demonstrate neurodevelopmental regression, involving language, sociability, play, and cognition (13). LKS represents selective loss of language in association with an abnormally paroxysmal EEG, eventually characterized by electrographic status epilepticus of slow-wave sleep (ESES).
While there is considerable overlap in the semiology of LKS and autism, some differences emerge. The great majority of children with autism who undergo language regression do so before three years of age (14), versus a mean age of language regression in LKS of 5–7 years. Only 10% of children with LKS regress before three years (4). As regression in autism occurs early, it usually entails the loss of single words, versus more drastic changes in LKS children who are typically older and have more developed vocabulary and language. LKS does not feature the behavioral profile that encompasses the core deficits of autism, i.e., abnormalities of reciprocal social relatedness and restricted stereotypical patterns of interests and behaviors. There is an intricate relationship between LKS, autism, ESES, and developmental dysphasias and the interaction between epileptiform discharges and cognitive dysfunction remains enigmatic (15). The presence of fluctuation in language and behavioral deficits, however, should raise concern regarding an accompanying diagnosis of epilepsy (13).
Focal epilepsy can interfere with language. Aphasic status epilepticus (16) and post-ictal aphasia (17) have been reported. Children with severe focal epilepsy from static brain lesions involving language cortex may have episodic ictal aphasia or status epilepticus and become permanently aphasic (3).
The early stages of LKS, with hyperkinesis and mild verbal auditory agnosia, may be confused with attention deficit hyperactivity disorder (ADHD) (10). Personality disturbances, aggressiveness, and depression are noted (18). Nonverbal developmental disorders may occur, but operational and intellectual capacities are usually preserved in LKS (9,19,20). The differential diagnosis also includes deafness, elective mutism, and acute psychiatric disorders.
Epileptic Manifestations
Seizures occur in approximately 70% of patients, one-third as a single seizure or episode of status, mostly at onset (21). In others, infrequent seizures occur between ages 5–10 years. After age 10, only one-fifth of patients continue with sporadic seizures; by age 15, seizures rarely persist. Seizures are often nocturnal simple partial motor, placing LKS on a spectrum that includes BECTS. Generalized tonic-clonic seizures, atypical absences, and myoclonic-astatic seizures occur less frequently (3). Complex partial seizures with psychomotor automatisms are rare (22). Tonic seizures are not characteristic (3,22). The frequency and type of seizures have no influence on prognosis. Treatment with anticonvulsant monotherapy is generally effective for seizure control, but not for the aphasia (3,9).
EEG Findings
Epileptiform abnormalities in LKS are variable, but striking. Bilateral independent temporal or temporoparietal spikes, bilateral 1-3 Hz slow-wave maximally temporal activity, generalized sharp- or slow-wave discharges, and multifocal or unilateral spikes are described (23). A report on spectral and topographic mapping of the EEG revealed variability in the mode of propagation of paroxymal discharges (24). Background activity is often normal or borderline. There is seldom activation by hyperventilation or photic stimulation. Epileptiform discharges are activated by sleep, especially sleep onset (Fig. 1). The significant activation of the spike-and-wave during non-REM sleep has led some to analogize to CSWS (21). Eventually in LKS, essentially all patients have bilateral spike-and-wave over 85% of non-REM sleep (ESES) (21). During a given night, however, these continuous discharges can be focal, restricted to the temporoparietal region (3). While nap recordings show the same abnormalities as those recorded during full nocturnal sleep, long-term EEG monitoring may be necessary to detect this phenomena (25).
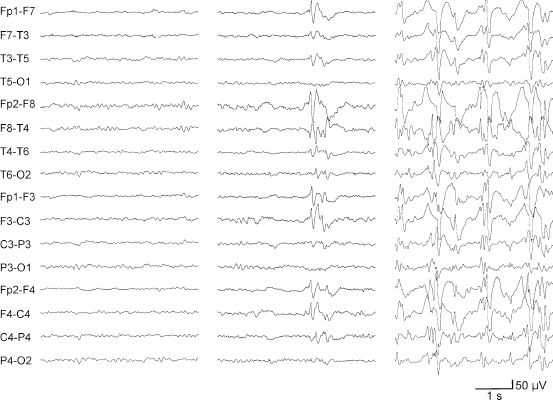
EEG in 5 year old girl with LKS. Note awake/drowsy background (left), activation of epileptiform activity in Stage 1 sleep (middle), and ESES in NREM sleep (right).
An interdependent relationship between language and epileptic manifestations has been described (26,27), though not all studies have suggested this (22). Holmes referred to the EEG abnormalities as epiphenomena of underlying pathology of cortex concerned with speech, rather than the cause of the aphasia (28). This view is supported by several observations: transient suppression of EEG discharges with intravenous benzodiazepines does not result in improvement of aphasia (21); EEG changes may not be accompanied by a change in aphasia (29); aphasia does not respond to conventional anticonvulsants despite seizure control (21,28); aphasia persists into adulthood despite normalization of EEG (9,20). Alternatively, improvement and worsening may coincide in the same direction, particularly in the sleep EEG (3,9). Disappearance of continuous spike-wave may herald improvement of aphasia (25). It seems reasonable to undertake treatment to terminate continuous EEG discharges early in LKS.
The placement of LKS on a spectrum with BECTS poses a challenge in the management of patients presenting with BECTS features who develop more ominous signs including neuropsychiatric dysfunction and medication refractoriness (30–32). Patients with otherwise classical seizures of rolandic epilepsy may develop atypical seizures, including generalized tonic-clonic, atonic, and atypical absences, as well as ESES and cognitive or behavioral disturbances. Attempts to distinguish EEG patterns specifically associated with LKS have identified unilateral slow wave foci, bilateral independent spike- and-wave discharges, and major activation of spike-and-wave discharges during sleep (33).
The interaction between LKS and autism poses another diagnostic dilemma. In a study of extended sleep EEGs in children with autism without epilepsy, an epileptiform EEG was identified in 14% of 155 children with regression, versus 6% of 364 without regression (34). Our study of 894 patients with autism spectrum disorder studied with overnight EEG revealed epileptiform potentials in 19% and no study revealed ESES (35). Overnight EEG appears warranted in autistic children with regression or fluctuation in their clinical course, but otherwise is not indicated in unselected patients.
Etiology
The etiology of LKS remains unknown, and may be due to diverse causes. Encephalitis has been postulated, but not verified (36). The clinical course is quite different from that of children with chronic encephalitis of the Rasmussen type. In a detailed report of two LKS cases that underwent partial temporal lobectomies, Cole et al. (37) from the Montreal Neurological Institute failed to uncover encephalitis. Pathological specimens were normal with the exception of mild subpial gliosis and occasional fibrous astrocytes throughout cortical gray matter. There was no evidence of inflammation, demyelination, hippocampal sclerosis, or dysgenesis. A report confirming encephalitic changes on cortical biopsy was given by Lou et al. (38). However, their case was atypical with elevated CSF protein and focal imaging abnormalities. Other biopsied cases did not have encephalitic changes (22,39). Angiography has occasionally suggested isolated arteritis of small and medium-sized vessels in some studies (40), but not others (22,37).
Other etiologies reported include a genetic predisposition (1,24,41), toxoplasmosis (42), neurocysticercosis (43,44), temporal astrocytoma (45), temporal ganglioglioma (46), hemophilus inflluenzae meningitis (19), subacute sclerosing panencephalitis (47), inflammatory demyelinating disease (48,49), and abnormal zinc metabolism (50). LKS may represent a final common pathway with multiple potential etiologies, acquired or genetic. The language disturbance, EEG abnormalities and epilepsy are likely the result of an insult to temporoparietal areas of the developing brain. As with other epilepsy syndromes, it would be useful to classify LKS into cryptogenic or symptomatic subgroups. Differences in pathogenesis, treatment, and prognosis may emerge, analogous to the infantile spasms of West syndrome.
Diagnostic Studies
Investigations have not delineated sufficient evidence to explain the pathophysiology. CSF (29,37,39,41), computed tomography (CT) (20,21,24,41,52), and magnetic resonance imaging (MRI) (21,41,54) findings are normal. There are uncommonly mild elevations of CSF protein (36,40,48), white matter changes on CT/MRI, or a structural lesion (43–46). Purists may argue that such cases represent other conditions mimicking LKS. Slight enlargement or asymmetry of the temporal horns have been reported (37,54), possibly secondary to the long-term epileptic process.
Various single photon emission computed tomography (SPECT) and positron emission tomography (PET) studies on small numbers of patients have shown temporal lobe abnormalities in brain perfusion and glucose metabolism (52,53,55). Chugani et al. (54) studied 17 LKS children with FDG-PET, demonstrating hypometabolism in the middle temporal gyri. Decreased temporoparietal perfusion has been seen with SPECT (55). A correlation between the aphasia of LKS with temporal lobe hypometabolism is not certain, as similar findings are observed in children with epilepsy who are not aphasic.
Treatment
The pharmacologic treatment of LKS is problematic due to several confounding observations. The benign course of the epilepsy versus devastating language impairment, fluctuating course of aphasia, lag of improvement in relation to the EEG, possibility of spontaneous remission, and rarity of the disorder render multiple barriers to controlled clinical trials. The determination of treatment efficacy is difficult. There is relatively scarce mention in the literature regarding antiepileptics of choice. Marescaux et al. (51) observed that phenobarbital, carbamazepine, and phenytoin were ineffective or even aggravating. Phenobarbital, having no effect on language, intensified behavioral problems, particularily hyperkinesis. Carbamazepine and phenytoin appeared to increase the duration of spike-wave activity in sleep. Valproate, ethosuximide, clonazepam, and clobazam were demonstrated to be partially or transiently effective. Clobazam has been reported to significantly reduce continuous spike-wave discharges in several small studies, associated with language improvement (27,51,56). Vigabatrin (57) and felbamate (58) have also been reported as effective.
Corticosteroids have been an efficacious treatment for both clinical and EEG abnormalities. This was reported by McKinney and McGreal (39), leading to the speculation of chronic encephalitis as the etiology of LKS. Effectiveness may be increased by early introduction (59). A recurrence of epileptiform EEG followed by an aphasic relapse has been described after tapering steroids (51). Prolonged, chronic, or intermittent therapy may be warranted if significant improvement of neuropsychological function is attained (3,51). Another recent addition is IVIG (49,60,61). The rationale for its use in LKS lies in the refractory nature of the epileptiform abnormalities, and the reports of beneficial effects in other intractable childhood epilepsies.
Surgical therapy including temporal lobectomy in lesional (45,46) and nonlesional (37) cases has been associated with improvement in language and seizure control. Multiple subpial transection (MST), designed to selectively disrupt intracortical horizontal fibers with minimal injury to vertically oriented cortical columns, has been suggested in treating epilepsies arising from eloquent or unresectable cortex. Morrell (62) reported a series of 14 LKS cases in which the epileptogenic discharges arose unilaterally, and were surgically treated with MST. Seven patients recovered age-appropriate speech; four showed marked improvement but continued in speech therapy programs; and the remaining three had no change. Other series have been small and demonstrated improvements that are often temporary (63), or in one series of five patients partial improvement but associated with a later extension of the procedure in one patient following relapse of ESES and clinical deficits (64). The most appropriate timing of this procedure and its long term ramifications are unknown. Further experience is needed to clarify the role of MST in the treatment of LKS.
Speech/language therapy is indispensable with periodic language and neuropsychological evaluations. Adverse behavioral manifestations may partly reflect frustration caused by aphasia. Introduction of an effective communication system could assist in alleviating such negative behavior. Some children with long standing verbal auditory agnosia are successfully integrated into schools for the deaf, although others continue to have marked deficits in social adaptation and communication. The patients who recover verbal language will drop the use of signs, allaying concern by some educators that there is detrimental competition between the two systems (3,65).
Impaired comprehension secondary to background noise in adolescents and adults who had recovered from LKS (9) suggests that improvement in the acoustic environment may enhance speech recognition ability. Listening may be assisted by increasing speech volume over ambient noise. This is accomplished with low-gain output personal or classroom FM systems or acoustically modifying the classroom (66). A comprehensive linguistic study of a 26-year old, left-handed male, with onset of LKS at age five years who learned sign language at age 13, revealed that sign language was the most efficient mode of communication. Severe restrictions in comprehension and production of spoken English or lip reading, and lesser impairment of reading, persisted. Functional MRI (fMRI) revealed strong activation of auditory cortex (R>L) to heard speech, little response to silent lip-reading, and strong activation of right temporo-parieto-occipital association cortex while viewing sign language (67). Further fMRI studies may provide an understanding of the extent of cortical impairment and applicability of various therapeutic strategies.
Prognosis
Several variables may influence prognosis, including age of onset, pattern of language deficit, frequency and topography of EEG discharges, duration of epilepsy, and efficacy, and adverse effects of anticonvulsants (3). There are few longterm follow up studies and no firm conclusions regarding potential for recovery (3,9,20). Outcomes range from complete recovery to permanent severe aphasia, with most experiencing improvement and residual moderate language deficits (68). In a study by Soprano et al. (69), no child with persistent EEG abnormalities recovered normal or near normal language; even among the nine whose EEG normalized; only three had complete recovery. A recent long term study of 11 patients with a mean follow-up of nine years eight months revealed complete language recovery in only 18.2% of cases and mental retardation in 63.6% (70). Adverse prognostic factors appear to be onset before 4 years, duration of aphasia longer than one year, and duration and continuity of ESES (4,70).
Conclusion
LKS is an epilepsy syndrome characterized by acquired aphasia and epileptiform EEG abnormalities eventually characterized by ESES. The language disorder could be the result of a paroxysmal disruption of language function during the time of its greatest development and vulnerability (71). In experimental animal models, there is clear evidence that functional disruption may interfere with the process of neuronal connection and cortical function (72). Seizures during a critical period for circuit development cause the emergence and fixation of permanent aberrant connections (73,74). LKS is a condition of unpredictable outcome and varying severity with a potentially relapsing remitting course, requiring constant adaptation and resourcefulness from parents, speech/language therapists, neuropsychologists, and neurologists.
Footnotes
Acknowledgments
The authors are grateful to Suzanne Reigle, B.S., and Kathleen Kelly, R.EEG T. for their assistance. This work was supported by a grant to GLH from the NINDS (NS27984) and a Mental Retardation Research Center Grant from NIH (HD18755-19).