
Abstract

Case report
An 18-year-old woman was hospitalized with severe headache associated with transient motor aphasia and right-sided sensory-motor deficit. Her past history was unremarkable, except for moderate, dull and fluctuant headache accompanied by a sore throat 3 weeks prior to admission. Twenty-four hours prior to hospitalization, her headache increased with nausea and vomiting. She further experienced a 1-h episode of numbness and weakness of her right proximal arm, spreading over 10 min to her right hand, followed by word-finding difficulties. On admission, she was alert, oriented and without fever or neck stiffness. Her neurological and neuropsychological examination was normal.
Routine blood tests were unremarkable. CSF analysis at admission revealed a lymphocytic pleocytosis with 350 WBC/mm3 and elevated protein at 2 g/L (Fig. 1A). Serologic tests were negative for syphilis, Lyme disease, brucellosis, mycoplasma pneumoniae and human immunodeficiency virus (HIV). There was no serological sign of recent infection for herpes simplex virus (HSV) 1-2, varicella zoster virus, Ebstein-Barr virus (EBV), rubella, measles or flavivirus. CMV IgM were slightly increased in the serum with no titre change over time and considered as unspecific by virologist. Serology for HHV-6 in the serum showed high titre of IgM that decreased to normal range over a period of 4 weeks (Fig. 1B). Polymerase chain reaction (PCR) in the CSF was negative for herpes 1-6. A repeated CSF analysis showed a persistent lymphocytic pleocytosis and high concentration of proteins that decreased dramatically over a period of 3 weeks (Fig. 1A). No CSF oligoclonal band was detected.
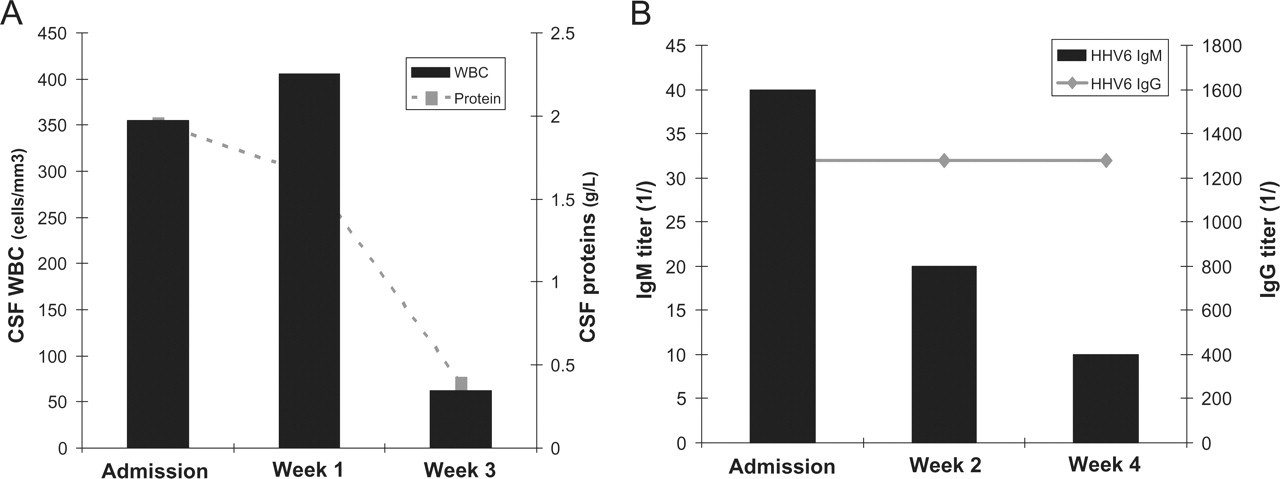
(A) CSF pleocytosis and protein content are markedly increased at admission and 1 week after. A normalization of CSF protein content and a dramatic decrease of CSF white blood cells are observed 3 weeks after admission. (B) HHV-6 serology: IgM are elevated at admission, decreasing by 50% 2 weeks after admission and back to normal range 4 weeks after admission. IgG titres are high but no variation is observed over the 4-week observation.
During the hospitalization, the patient experienced eight episodes of transient, self-limiting neurological deficits lasting 60 to 180 min. These episodes were either preceded or followed by severe throbbing headache, sometimes associated with vomiting. These neurological deficits were characterized by speech difficulties (either word-finding difficulties or severe dysarthria) and fluctuating sensory motor symptoms involving the right arm, both hands, or the perioral region and the tongue. There was no loss of consciousness and the patient later remembered these episodes. Between those episodes, the patient was asymptomatic and her neurological examination was normal.
An acute episode was recorded during a 24-h video-EEG monitoring (Fig. 2A–C). Twenty minutes after onset, the patient presented with dysarthria and numbness of the hands, perioral area and mouth; EEG revealed non-rhythmic slow waves in the left temporo-parieto-occipital region. Two hours after, EEG showed a diffuse bilateral slowing. Cerebral MRI performed before the first spinal tap revealed diffuse leptomeningeal gadolinium enhancement of the brain and was otherwise normal (Fig. 3A–C). Cranial CT phlebography, MRI of the spinal cord and CT of the thorax were normal.
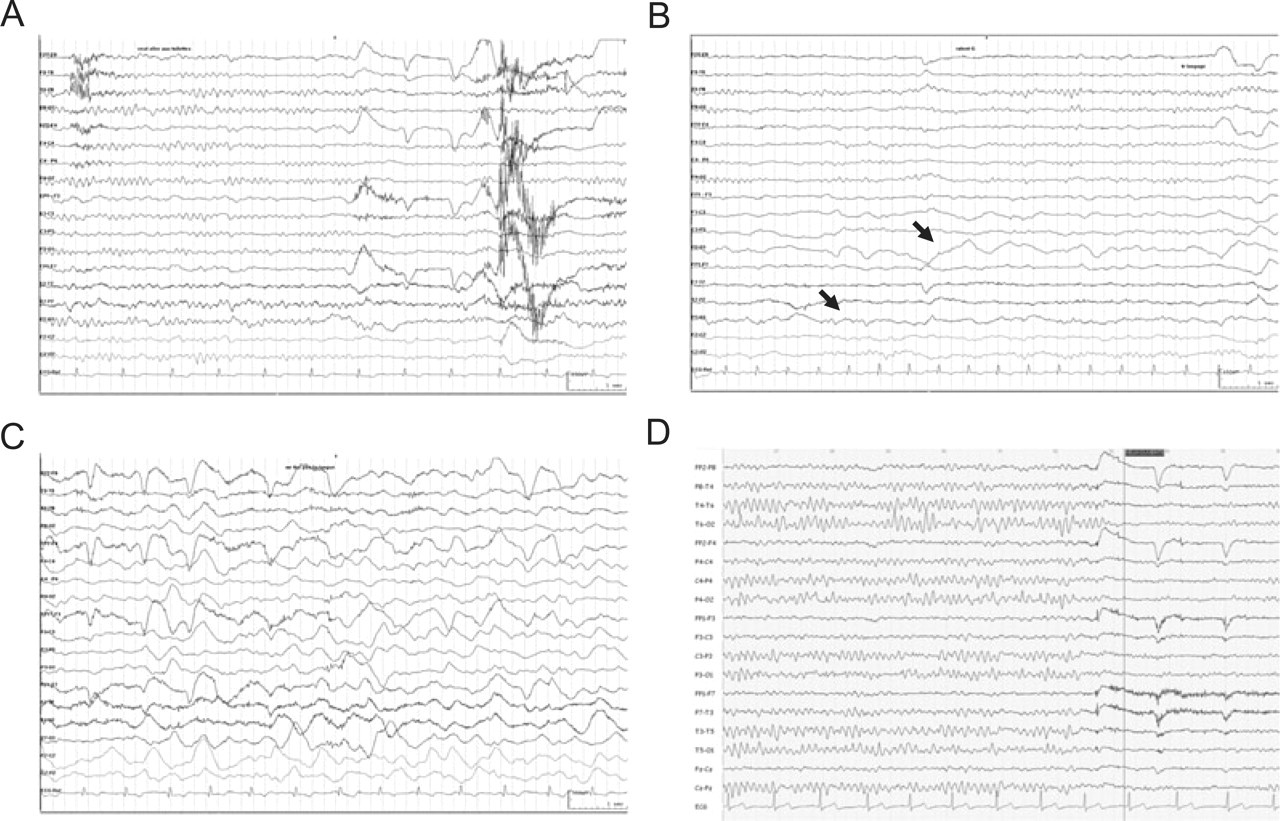
Long-term EEG (high pass 1 Hz, low pass 70 Hz, 10 s/page). (A) Onset of an acute episode: wakes up, headache, normal background, no focal slowing. (B) During neurological deficit (20 min after onset): important left posterior (temporo-parieto-occipital) delta slowing (arrows) with disappearance of the physiological alpha rythm. (C) Persisting acute episode (2 h after onset): bilateral diffuse slowing. (D) Nine months after the episode: normal background, no focal slowing.
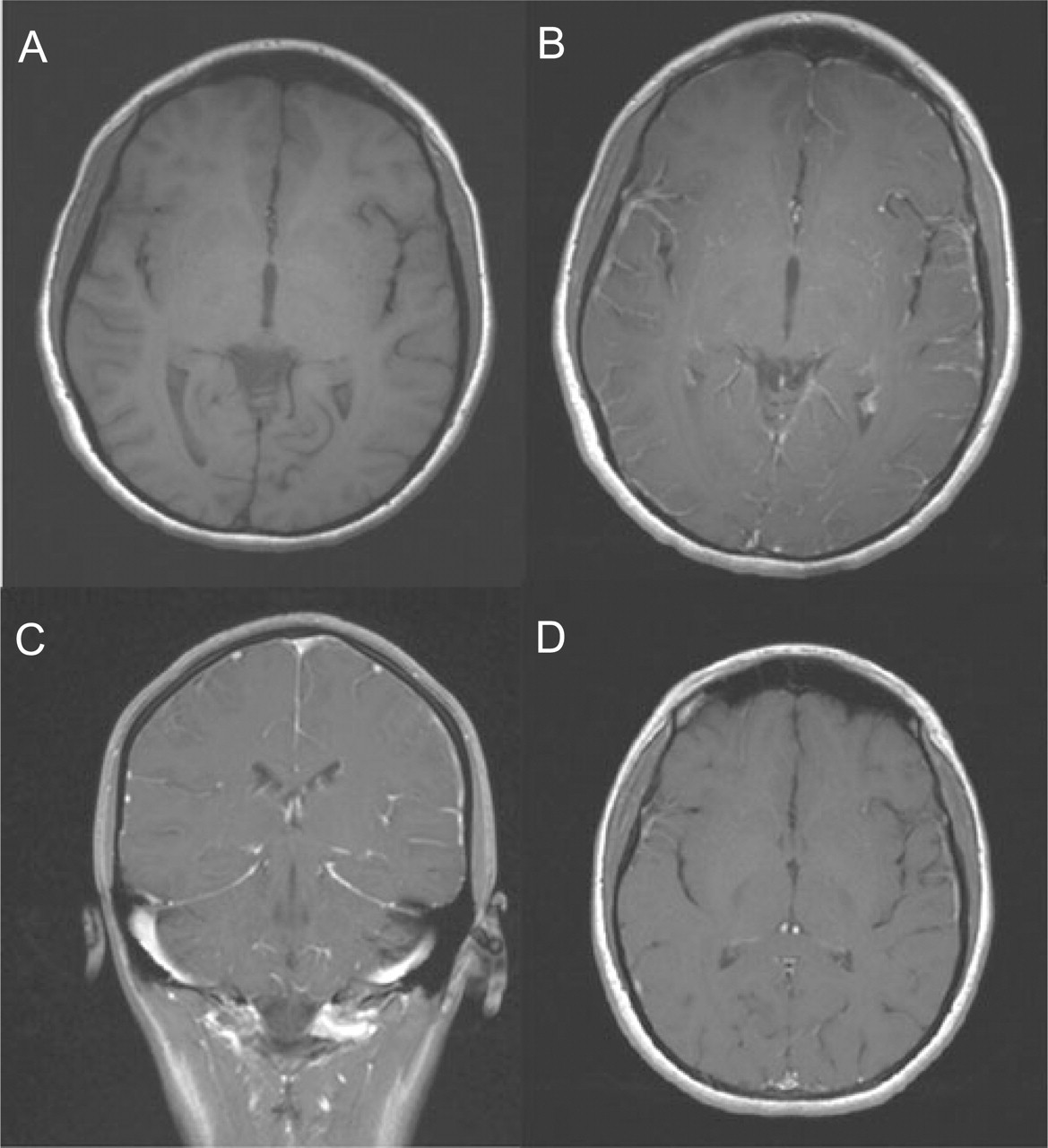
Brain MRI at admission: axial T1-weighted without (A) and with (B) gadolinium and coronal T1-weighted with gadolinium (C) showing diffuse leptomeningeal enhancement. Control brain MRI 1 month after admission: axial T1-weighted with gadolinium shows disappearance of leptomeningeal enhancement (D).
The patient was treated with acyclovir (10 mg/kg/8 h) and ceftriaxone (2 g/12 h). Ceftriaxone was stopped after 3 days and acyclovir was continued for a total of 14 days. During hospitalization the patient remained afebrile. She presented her last transient neurological deficit 5 days after acyclovir discontinuation. Upon follow-up 1, 3 and 9 months after her discharge, the patient was asymptomatic. Control brain MRI performed 4 weeks after admission showed the disappearance of leptomeningeal gadolinium enhancement and was otherwise normal (Fig. 3D). Follow-up EEG 9 months after the episode showed complete resolution of the abnormalities (Fig. 2D).
Discussion
The syndrome of transient headache and neurological deficits with CSF lymphocytosis (HaNDL syndrome) (1, 2), also known as ‘Pseudomigraine with lymphocytic pleocytosis and transient neurological deficits’ (PMP syndrome) (3), is described as a benign and self-limiting neurological illness mainly seen in young and middle-age adults. This syndrome is associated with several episodes of headache accompanied by transient neurological deficits and is usually preceded by viral prodromes and resolves completely in less than 3 months. The recurrent neurological deficits typically last no more than 3 days, with no signs or symptoms between episodes. In addition, CSF analysis shows lymphocytic pleocytosis with increased total protein amount. Paraclinical tests are usually normal except for single photon emission computed tomography (SPECT) of the brain and EEG that can show abnormalities.
Our patient's neurological symptoms fulfil diagnostic criteria for HaNDL syndrome (1, 3). The onset of disease was preceded by virus-like symptoms and recurrent headache was associated with transient neurological symptoms separated by asymptomatic periods. CSF examination was characteristic for HaNDL syndrome with CSF lymphocytic pleocytosis associated with a substantial increase of proteins within the CSF (2, 3). The EEG showed focal slowing compatible with HaNDL (4).
Her presentation is noteworthy for two reasons. First, she had a typical course of HaNDL with leptomeningeal enhancement on cerebral MRI, not reported hitherto. Second, the combination of headache and sore throat starting 3 weeks prior to hospitalization, together with serological evidence of recent HHV-6 infection, suggests that she had been subject to a recent infection with a virus of the herpes family.
The aetiology of HaNDL syndrome is unknown and it has been hypothesized that it may result from immune-mediated mechanisms after a viral infection (2, 3). The significance of the CSF pleocytosis has been attributed to a non-infectious central nervous system (CNS) inflammation secondary to migraine (4, 5), while others have suspected a consequence of viral meningitis (6, 7). In a large study including 50 patients suffering from HaNDL syndrome, no laboratory evidence for recent infection including herpes 1 to 3 (herpes simplex virus (HSV)-1/2, varicella zoster virus (VZV)) and 5 (cytomegalovirus (CMV)) was detected (3).
To the best of our knowledge, HHV-6 infection has not been assessed in studies on HaNDL syndrome. CMV infection has been associated in a single case report of HaNDL (8), but the absence of positive CMV in a large study including 50 patients with HaNDL syndrome could not confirm this hypothesis. Other reports suggested a possible association with borrelia burgdorferi, HSV 1-2, echovirus, but were not further confirmed (3).
HHV-6 belongs to the β-herpes family and is expressed in the form of two variants, A and B (9, 10). These subtypes share a high level of sequence homology, but differ in their phenotype. HHV-6B, on the one hand, is acquired early in life, and causes roseola (9). The vast majority (> 95%) of the general population has been exposed to HHV-6B and carries the virus latently. On the other hand, HHV-6A is not linked to any clearly defined syndrome yet. HHV6 infection can occasionally result in encephalitis in immunocompetent adults (11). Thus, it is possible to be infected by a single variant of HHV-6 during childhood and by the other variant, sporadically, during adulthood. Unfortunately, serological assay cannot distinguish the two variants, which can be done only by PCR.
Because 25–40% of patients with HaNDL syndrome have symptoms of a preceding viral illness (3), it is conceivable that viruses are implicated in the pathogenesis of HaNDL either directly or through mechanisms of molecular mimicry (9). The serological constellation of positive IgM antibodies against HHV-6 with titre change of four times over a period of 4 weeks observed in our patient support a recent infection, although a virus reactivation in situ cannot be excluded. The involvement of virus in this case is further supported by the demonstration of radiological leptomeningeal gadolinium enhancement, as typically seen after viral meningitis. The absence of positive PCR for HHV-6 in the CSF can be explained by the low sensitivity of the assay as well as the time course of the sampling. In this case, the spinal tap was performed 3 weeks after symptoms onset and this delay may explain the negative PCR result. Indeed, it must be kept in mind that PCR performed to detect virus DNA may give false-negative results when a CSF sample is taken either within the first 3 days or more than 14 days after viral illness onset (12), as was the case in our patient. HaNDL syndrome typically runs a self-limiting course with spontaneous resolution of symptoms (2, 3), which is in line with the excellent outcome witnessed in our patient. This case shows no direct evidence that treatment with acyclovir was beneficial, although we cannot exclude that it contributed to her rapid and favourable clinical evolution. Because intravenous acyclovir is not without risks, these data suggest an area for future work.
In conclusion, while the aetiology of HaNDL remains obscure, our observation supports the hypothesis that in some cases, HHV-6 may trigger the HaNDL syndrome either directly or via a post-infectious mechanism. Further studies assessing the role of HHV-6 in HaNDL syndrome are needed.
Footnotes
Acknowledgements
The authors want to thank Dr Luc Perrin (Virology, University Hospital of Geneva) for helpful discussion and Dr Maria-Isabel Vargas (Neuroradiology, University Hospital of Geneva) for the selection of MRI images.