
Abstract

Familial hemiplegic migraine (FHM) is a rare and severe autosomal-dominant subtype of migraine with aura (MA) in which attacks are associated with some degree of hemiparesis in addition to other neurological aura symptoms (1). Regarding the frequency and severity of attacks, there is a high degree of phenotypic variability—both inter- and intraindividually. The aura phase can last from several hours to days or even weeks. Other complications during the attacks include fever (with or without cerebrospinal fluid pleocytosis), confusion, impaired consciousness, coma and epileptic seizures.
Genetically, FHM is heterogeneous. Mutations in three different genes have been identified in affected pedigrees: CACNA1A (FHM1; OMIM 141500), encoding the pore-forming α1a subunit of a neuronal, voltage-gated, P/Q-type calcium channel (2, 3), ATP1A2 (FHM2; OMIM 602481), encoding an astrocytic sodium-potassium pump (4), and SCN1A (FHM3; OMIM 609634), encoding the α subunit of a voltage-gated neuronal sodium channel (5, 6). More than 50% of FHM pedigrees are linked to the FHM1 CACNA1A locus, making CACNA1A the most frequent FHM gene. So far, more than 15 missense mutations associated with FHM have been published, the T666M mutation in exon 16 being the most frequent one.
Alhough primarily a paroxysmal disorder, FHM can be complicated by permanent neurological symptoms in the interval between attacks, the most frequent one being a cerebellar syndrome (FHM with ataxia) (2). In addition, mental retardation (MR) has been recognized as part of the phenotypic spectrum. Although observed in several families with FHM2 (4, 7, 8), it has been reported only sporadically in FHM1 (9–11), and it remained unclear whether this was more than a chance co-occurrence.
Here, we present clinical, neuropsychological and genetic data of a large German FHM1 family with the previously reported T666M CACNA1A mutation, in which several individuals suffered from early-onset permanent MR in addition to typical attacks of FHM, suggesting that MR can be part of the phenotypic spectrum not only of FHM2, but also of FHM1.
Case report
We describe a three-generational German FHM family (Fig. 1). All probands were personally examined and underwent a detailed semistructured interview (45–90-min duration), which included questions about migraine, other types of headache, epileptic seizures and general past medical history (5). Diagnosis of FHM was established according to the criteria of the International Headache Society (1).

Co-segregation of the T666M mutation with the phenotype within the pedigree. Circle, female; square, male; arrow, index patient; upper half black, affected by hemiplegic migraine; lower left quarter black, mentally retarded; lower right quarter black, cerebellar syndrome; question mark, phenotype unknown; numbers next to symbols, identifiers used in the text and the tables; +, presence of mutation; –, absence of mutation; o, DNA not available. Comments: In III:17, information on mental retardation (MR) and cerebellar dysfunction is missing, since he was not available for examination and interview. In III:11 there was no evidence of MR, although formal neuropsychological testing could not be performed.
A total of four individuals (II:4, II:6, II:8, III:11) suffered from typical hemiplegic attacks. In all of them, frequency, duration and severity of attacks were at the lower end of the phenotypic spectrum, without occurrence of unusual aura features such as confusion, fever, epileptic seizures or coma (Table 1). All of them displayed signs of mild (II:4, III:11) to severe (II:6, II:8) cerebellar dysfunction in the interval between attacks, with evidence of cerebellar vermian atrophy on cMRI in two of them [II:6, II:8].
Phenotypic features of CACNA1A mutation carriers

For definition of MR refer to Table 2.
Was hospitalized for several weeks because of confusional state of unknown aetiology, but there was not any correlation with attacks of hemiplegic migraine.
Bilateral motor deficits on rare occasions.
Not available for neuropsychological testing.
Did not experience any hemiplegic attacks or other types of migraine.
HM, Hemiplegic migraine; HA, headache; H, hemiparesis/hemiplegia; S, sensory disturbances; V, visual disturbances; A, aphasia; +, indicates that respective symptom was consistently present in all/most attacks; –, indicates that respective symptom was never present; TR, temporal relationship between aura and HA phase (A > HA indicates that HA starts after aura); HA : ND sides, relationship between localization of HA and side of neurological deficit(s); N/V/P/P, nausea/vomiting/photophobia/phonophobia; Szs, epileptic seizures; TTH, tension-type headache; n.a., not applicable; n.d., not determined.
III:17 was not available for direct examination and interview, but was reported to suffer from regular hemiplegic attacks. III:14 did not experience any hemiplegic attacks; except for an episode of reduced consciousness of a few hours' duration without accompanying headache or hemiparesis at the age of seven years, his past medical history was unremarkable. However, he showed a mild cerebellar syndrome, with cerebellar vermian atrophy on cerebral magnetic resonance imaging (cMRI).
In four affected individuals (II:4, II:6, II:8, III:14) detailed neuropsychological testing was performed (for details of the test batteries and definition of MR refer to Table 2). Interestingly, the proband (II:6) and two other individuals (II:8 and III:14) were found to be mentally retarded, with IQs of 70, 76 and 80, respectively (Tables 1 and 2). All of them failed to finish school and worked in unskilled manual labour. As far as could be assessed retrospectively, there was no evident causal relationship between the course of FHM attacks and cognitive dysfunction in II:6 and II:8, e.g. a stepwise deterioration of MR after recurrent or severe attacks of FHM. Age at onset (AaO) for MR was in early childhood in all patients, i.e. prior to the onset of hemiplegic attacks.
Results of neuropsychological testing
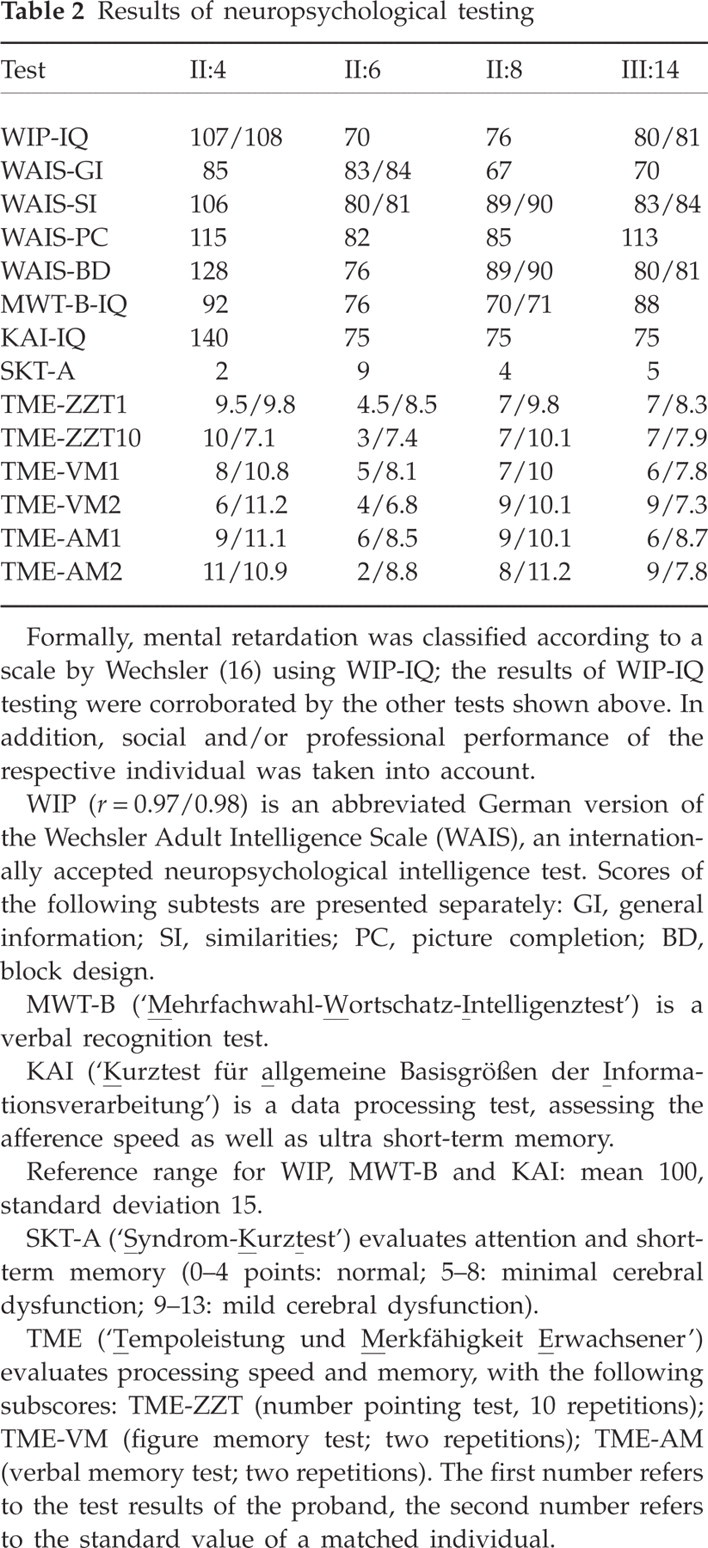
Formally, mental retardation was classified according to a scale by Wechsler (16) using WIP-IQ; the results of WIP-IQ testing were corroborated by the other tests shown above. In addition, social and/or professional performance of the respective individual was taken into account.
WIP (r = 0.97/0.98) is an abbreviated German version of the Wechsler Adult Intelligence Scale (WAIS), an internationally accepted neuropsychological intelligence test. Scores of the following subtests are presented separately: GI, general information; SI, similarities; PC, picture completion; BD, block design.
MWT-B (‘
KAI (‘
Reference range for WIP, MWT-B and KAI: mean 100, standard deviation 15.
SKT-A (‘
TME (‘
Venous EDTA blood samples were obtained from all available family members (five affected, one unaffected), and DNA was extracted from peripheral leukocytes according to standard techniques. In the index patient (II:6), all exons as well as exon–intron boundaries of the CACNA1A gene were analysed for mutations (5). The previously described CACNA1A T666M mutation (c.C2233T; AF004884) was identified; it showed complete co-segregation with the FHM phenotype, without phenocopies (Fig. 1). Of note, T666M was also detected in III:14, who did not suffer from hemiplegic migraine, but showed cerebellar dysfunction and MR (reduced penetrance).
Chromosome analysis (450 bands/GTG-banding) revealed a normal karyotype in the index patient (II:6). In addition, fragile X syndrome was ruled out in the index patient by molecular genetic analysis of the FRAXA gene, which showed a normal number (n = 29) of CGG repeats (polymerase chain reaction and Southern blot technology).
Discussion
We describe a German FHM family in which the previously reported T666M mutation in CACNA1A was identified. Of note, the proband (II:6) and two other mutation carriers (II:8 and III:14) suffered from permanent MR in addition to the FHM phenotype.
Several lines of evidence support the notion that MR in this family is part of the phenotypic spectrum of FHM rather than representing a chance co-occurrence: (i) two frequent causes of MR, chromosomal abnormalities and fragile X syndrome, were ruled out; (ii) a likelihood ratio test, comparing the likelihood that MR is co-segregating independently vs. the likelihood that MR is part of the phenotypic spectrum of FHM, indicated a positive trend (although not significant due to limited power) towards the hypothesis of MR being part of the phenotypic spectrum of FHM; (iii) MR has been observed in episodic ataxia type 2 (EA2), a disorder which is allelic to FHM1 and also caused by mutations in CACNA1A (12); (iv) MR has been reported sporadically in FHM, although the data hardly allowed a conclusion, if MR is more than a chance association.
There are several reports of MR in pedigrees in which the exact FHM subtype is unknown, since the diagnosis was not genetically confirmed (13, 14). Apart from these reports, MR has been predominantly reported in FHM2 pedigrees; it has been observed for a total of seven FHM patients who were carriers of the G615R, D718N, P979L, L746P mutations in the ATP1A2 gene (4, 7, 8). More importantly, MR has also been described in a total of five CACNA1A mutation carriers. Vahedi et al. have reported a sporadic patient suffering from severe attacks of hemiplegic migraine (with coma and seizures) and early-onset profound cognitive dysfunction (estimated IQ of 40), in whom a de novo Y1385C mutation in CACNA1A was identified (11). Kors et al. (2001) have described a previously published (15) family with FHM1 caused by the CACNA1A S218L mutation, in which the affected brother of the proband was found to be ‘mildly mentally retarded’; however, AaO and severity of MR were not further specified (10). Finally, and most importantly, Kors et al. (2003) have reported on a British FHM1 family with the CACNA1A T666M mutation, in which three mutation carriers were found to be mentally retarded (9). Again, the precise AaO as well as the severity of MR was not explicitly stated.
Taking all data together, MR has been observed in a few FHM1 patients; of note, all of them were suffering from FHM plus cerebellar ataxia, similar to the affected individuals in our family.
Against this background it is highly likely that the MR observed in three individuals in our family is causally related to the CACNA1A genotype. This raises the fundamental question whether MR occurs secondary to neuronal damage caused by repeated attacks of FHM, or is caused by permanent neuronal damage due to dysfunction of the CACNA1A calcium channel.
Several aspects of the clinical history of our family argue in favour of the latter scenario: (i) anamnestically, AaO for MR was in early childhood in all individuals, whereas attacks of FHM started during youth, i.e. with a latency of several years. This was corroborated by results of neuropsychological testing which were compatible with early-onset cognitive dysfunction; (ii) one mentally retarded individual (III:14) did not have a single attack of hemiplegic migraine in his lifetime; (iii) no particularly severe attacks with either prolonged duration or complicating features such as confusion or reduced consciousness were observed in any of the mentally retarded individuals; (iv) as far as could be assessed retrospectively, MR did not develop in a stepwise fashion, with gradual deterioration after attacks of FHM.
Of note, these aspects are in contrast to the FHM1 family with MR reported by Kors et al. (2003), in which a stepwise cognitive decline was observed, with worsening after each attack (9). This leaves the possibility that both pathogenic mechanisms, i.e. permanent neuronal damage due to calcium channel dysfunction and neuronal damage as a result of repeated attacks of FHM, have a combined effect in causing the MR phenotype.
Although the prognosis of FHM is generally favourable, the family presented here, together with other previously published pedigrees, shows that FHM is a complex syndrome which can be associated with permanent deficits such as cognitive dysfunction in some patients. Of note, this is true not only for the FHM2 subtype, but also for FHM1 associated with CACNA1A mutations; thus, the frequently propagated notion that MR is more common in FHM2 families has to be revised. Although MR has previously been observed in sporadic patients with CACNA1A mutations (11) and single individuals from FHM1 families (10), to our knowledge this is only the second report of a FHM1 family, in which several affected family members displayed marked MR (9). In contrast to previous reports of FHM1 associated with MR in which affected individuals showed a particularly severe FHM phenotype, our data emphasize that MR can occur also in the setting of ‘regular’ attacks of FHM without complicating features. Thus, screening for cognitive dysfunction should be part of the standard phenotyping protocol in hemiplegic migraine—both in clinical routine and in research.
Based on our observations, permanent MR in the setting of FHM seems to share several features with the other major permanent deficit associated with FHM, i.e. cerebellar ataxia: (i) both seem to have an early onset without pronounced increase in severity over time; (ii) both can occur in mutation carriers without particularly severe hemiplegic attacks or without hemiplegic attacks at all.
In our family, the same CACNA1A T666M mutation causes a spectrum of different phenotypes ranging from hemiplegic migraine alone (II:4, III:11), hemiplegic migraine plus MR (II:6, II:8) to MR without recurrent attacks of hemiplegic migraine (III:14). This variability is probably due to environmental factors and/or modifier genes yet to be identified.